Tutorials_scCAMEL-SWAPLINEv1_LiverMacrophage
Original Article: Human resident liver myeloid cells protect against metabolic stress in obesity,”Nature Metabolism.”, 2023
Package: scCAMEL-SWAPLINE.v1
[11]:
import datetime
today=f"{datetime.datetime.now():%Y-%m-%d}"
today
[11]:
'2023-07-07'
[2]:
import torch
import torch.nn as nn
from torch.autograd import Variable
import torch.utils.data as Data
import torchvision
import matplotlib.pyplot as plt
from mpl_toolkits.mplot3d import Axes3D
import torch.utils.data as data_utils
from matplotlib import cm
import numpy as np
import pandas as pd
import pickle as pickle
from scipy.spatial.distance import cdist, pdist, squareform
import pandas as pd
from sklearn.linear_model import LogisticRegression, LogisticRegressionCV
from sklearn.model_selection import StratifiedShuffleSplit
from collections import defaultdict
from sklearn import preprocessing
import matplotlib.patches as mpatches
import torch.nn.functional as F
import math
#import gpytorch
import urllib.request
import os.path
from scipy.io import loadmat
from math import floor
import anndata
# Make plots inline
%pylab inline
%pylab is deprecated, use %matplotlib inline and import the required libraries.
Populating the interactive namespace from numpy and matplotlib
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\IPython\core\magics\pylab.py:162: UserWarning: pylab import has clobbered these variables: ['floor']
`%matplotlib` prevents importing * from pylab and numpy
warn("pylab import has clobbered these variables: %s" % clobbered +
[3]:
torch.manual_seed(1) # reproducible
[3]:
<torch._C.Generator at 0x24082a98eb0>
[4]:
import scCAMEL as scm
from scCAMEL import CamelPrefiltering
from scCAMEL import CamelSwapline
from scCAMEL import CamelEvo
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scCAMEL\CamelSwapline.py:534: FutureWarning: The default dtype for empty Series will be 'object' instead of 'float64' in a future version. Specify a dtype explicitly to silence this warning.
def addcolor(datax,clustername="Cluster", colorcode="color", predef=pd.Series()):
[26]:
screfall=anndata.read("LiverMacrophage_474cells_Ref2023-01-16_MergeCluster_35epch.h5ad")
screfall
[26]:
AnnData object with n_obs × n_vars = 474 × 21397
obs: 'cellID', 'Cluster'
[28]:
set(screfall.obs["Cluster"])
[28]:
{'LM1', 'LM2-C1', 'LM2-C2', 'LM3', 'LM4'}
[29]:
scref=screfall
[30]:
set(scref.obs["Cluster"])
[30]:
{'LM1', 'LM2-C1', 'LM2-C2', 'LM3', 'LM4'}
[31]:
scref.obs.groupby(["Cluster"]).count()
[31]:
| cellID | |
|---|---|
| Cluster | |
| LM1 | 133 |
| LM2-C1 | 39 |
| LM2-C2 | 73 |
| LM3 | 96 |
| LM4 | 133 |
[35]:
scref.X=scref.X.todense()
[36]:
path='/Dropbox/data/proj/PE_HYZ/PublicDataSet/'
filename='PANTHER_cell_cycle_genes.txt'
#dfpfc2= prefilteringTest.prefilter(df_f=dfpfc,filename=filename, path=path)
#scref= scm.CamelPrefiltering.prefilter(datax=scref,filename=filename, path=path)
[37]:
scref=scm.CamelPrefiltering.DataScaling(scref)
[38]:
scref.var['Filter1']=[True]*scref.var.shape[0]
scref
[38]:
AnnData object with n_obs × n_vars = 474 × 21397
obs: 'cellID', 'Cluster'
var: 'Filter1'
[43]:
scref=CamelPrefiltering.SelectFeatures(datax=scref, clustername='Cluster',methodname='wilcoxon', numbergenes=1000, folderchange=1.5)
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: overflow encountered in expm1
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:418: RuntimeWarning: overflow encountered in expm1
self.expm1_func(mean_rest) + 1e-9
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: invalid value encountered in true_divide
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:420: RuntimeWarning: divide by zero encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: overflow encountered in expm1
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:418: RuntimeWarning: overflow encountered in expm1
self.expm1_func(mean_rest) + 1e-9
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: invalid value encountered in true_divide
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:420: RuntimeWarning: divide by zero encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: overflow encountered in expm1
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:418: RuntimeWarning: overflow encountered in expm1
self.expm1_func(mean_rest) + 1e-9
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: invalid value encountered in true_divide
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:420: RuntimeWarning: divide by zero encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: overflow encountered in expm1
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:418: RuntimeWarning: overflow encountered in expm1
self.expm1_func(mean_rest) + 1e-9
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: invalid value encountered in true_divide
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:420: RuntimeWarning: divide by zero encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: overflow encountered in expm1
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:418: RuntimeWarning: overflow encountered in expm1
self.expm1_func(mean_rest) + 1e-9
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:417: RuntimeWarning: invalid value encountered in true_divide
foldchanges = (self.expm1_func(mean_group) + 1e-9) / (
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scanpy\tools\_rank_genes_groups.py:420: RuntimeWarning: divide by zero encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
[45]:
########################################################
########################################################
#remeber to change the file path in tftable
########################################################
########################################################
scref =scm.CamelPrefiltering.LabelGene_Scaling(datax=scref,
TPTT=100000, mprotogruop=scref.obs["Cluster"].values,commongene=None,
sharedMVgenes=None,std_scaling=True,
tftable="(file path to tftable)/FantomTF2CLUSTER_human_official.txt", learninggroup="train")
CamelRunning---GenesScaling......
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scCAMEL\CamelPrefiltering.py:576: FutureWarning: In a future version of pandas all arguments of DataFrame.dropna will be keyword-only.
scalepfc = dfpfc.div(dfpfc.std(1), axis=0).dropna(0)
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scCAMEL\CamelPrefiltering.py:577: FutureWarning: In a future version of pandas all arguments of DataFrame.dropna will be keyword-only.
scalepfc = dfpfc.astype(float).dropna(0)
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scCAMEL\CamelPrefiltering.py:579: FutureWarning: In a future version of pandas all arguments of DataFrame.dropna will be keyword-only.
scalepfc = scalepfc.dropna(0)
C:\Users\huyiz\AppData\Local\Continuum\anaconda3\envs\py39\lib\site-packages\scCAMEL\CamelPrefiltering.py:580: FutureWarning: Passing a set as an indexer is deprecated and will raise in a future version. Use a list instead.
dfpfc_dev = scalepfc.loc[set(scalepfc.index) & set(sharedMVgenes)].dropna()
CamelRunning---TrainingGenesScaling......Finished
[46]:
scref
[46]:
AnnData object with n_obs × n_vars = 474 × 21397
obs: 'cellID', 'Cluster', 'mtrain_index'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'rank_genes_groups', 'train_set_gene', 'mclasses_names'
obsm: 'train_set_values'
[47]:
len(scref.var.index[scref.var["MVgene"]])
[47]:
3987
[48]:
net=scm.CamelPrefiltering.NNclassifer(
datax=scref,
epochNum=200,
learningRate=0.03,
verbose=0,
optimizerMmentum=0.8,
dropout=0.3,
#imizer__nesterov=True,
)
CamelRunning---NNclasffier_in_cuda.......
CamelRunning---NNclasffier_in_cuda.......Finished
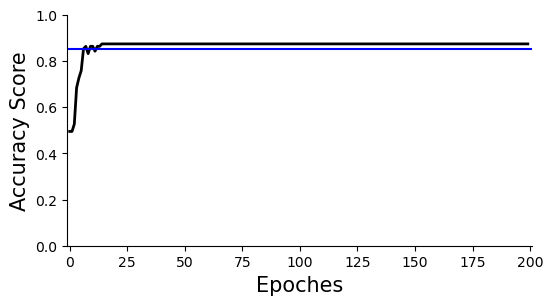
#Accuracy plot, the overall clustering accuracy is ~85%
[52]:
ax=scm.CamelPrefiltering.AccuracyPlot( nnModel=net, accCutoff=0.85,
Xlow=-1, Ylow=0.0, Yhigh=1,
)
plt.savefig("upload_%s_CurvePlot_learningAccuracy.pdf"%today,bbox_inches='tight')
[53]:
net=scm.CamelPrefiltering.NNclassifer(
datax=scref,
epochNum=35,
learningRate=0.03,
verbose=0,
optimizerMmentum=0.8,
dropout=0.3,
#imizer__nesterov=True,
)
CamelRunning---NNclasffier_in_cuda.......
CamelRunning---NNclasffier_in_cuda.......Finished
[54]:
scref
[54]:
AnnData object with n_obs × n_vars = 474 × 21397
obs: 'cellID', 'Cluster', 'mtrain_index'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'rank_genes_groups', 'train_set_gene', 'mclasses_names'
obsm: 'train_set_values'
[56]:
#if color is not defined
scref=scm.CamelSwapline.addcolor(datax=scref,clustername="Cluster", colorcode="color")
[57]:
scref
[57]:
AnnData object with n_obs × n_vars = 474 × 21397
obs: 'cellID', 'Cluster', 'mtrain_index', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'rank_genes_groups', 'train_set_gene', 'mclasses_names', 'refcolor_dict'
obsm: 'train_set_values'
[58]:
set(scref.obs["Cluster"])
[58]:
{'LM1', 'LM2-C1', 'LM2-C2', 'LM3', 'LM4'}
[ ]:
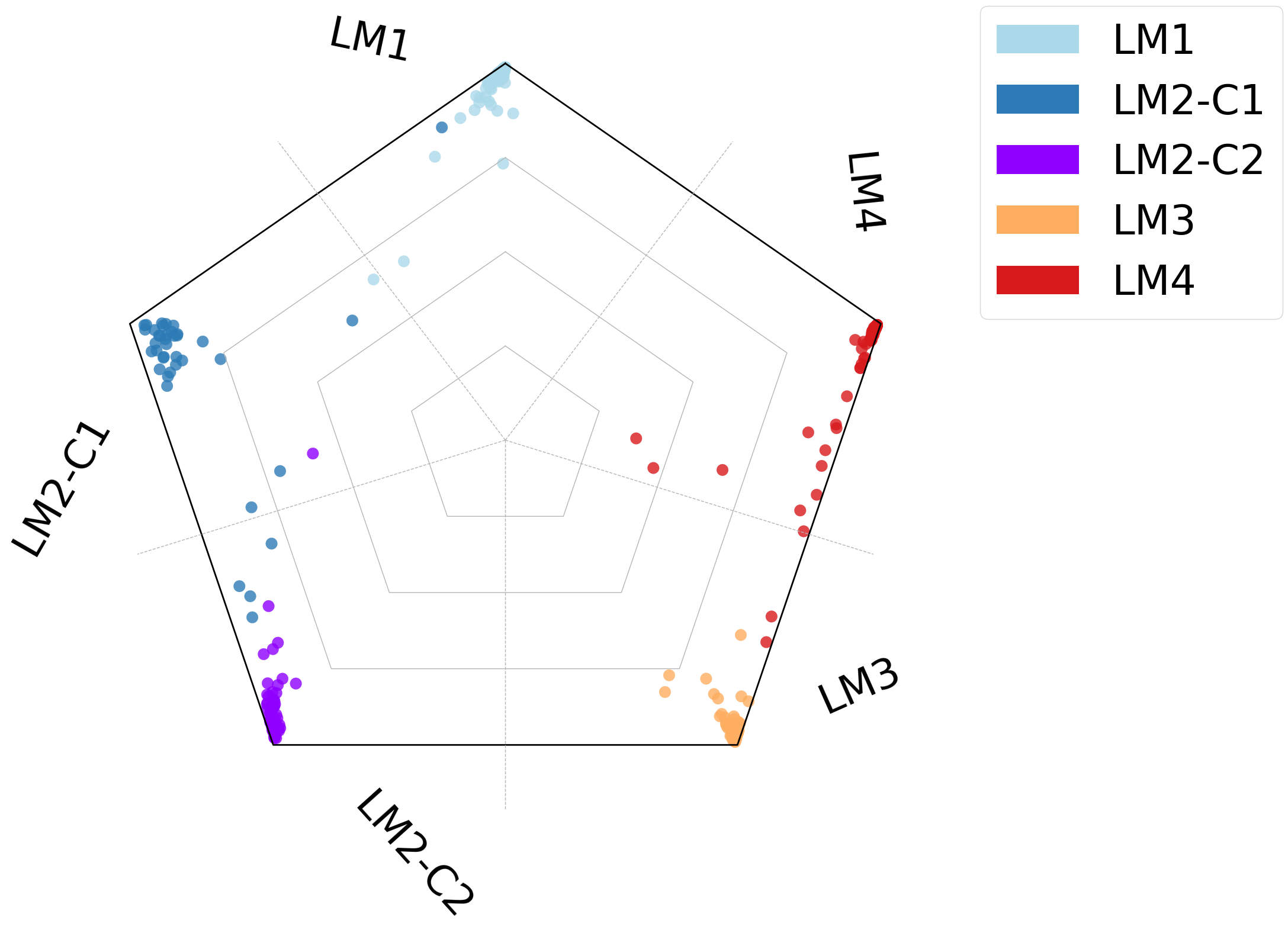
LM1: #ABD9E9
LM2-C1 (use name LM2): #2C7BB6
LM2-C2 (use name cDC2): purple
LM3: #FDAE61
LM4: #D7191C
[59]:
clist=[]
for item in scref.obs["Cluster"]:
if item=="LM1":
clist.append("#ABD9E9")
elif item=="LM2-C2":
clist.append("#2C7BB6")
elif item=="LM2-C1":
clist.append("purple")
elif item=="LM3":
clist.append("#FDAE61")
elif item=="LM4":
clist.append("#D7191C")
[60]:
scref.obs["color"]=clist
[61]:
scref.uns['refcolor_dict']={'LM2-C1': [44, 123, 182],
'LM2-C2': [143, 0, 255],
"LM3": [253,174,97],
"LM4": [215,25,28],
'LM1': [171, 217, 233],
}
[62]:
scref.uns["mwanted_order"] =list(sort(list(set(scref.obs["Cluster"]))))
[64]:
scref=scm.CamelSwapline.prediction(datax=scref, mcolor_dict=scref.uns["refcolor_dict"] ,net=net,learninggroup="train", radarplot=True,fontsizeValue=18,
ncolnm=3, bbValue=(1.2, 1.05) )
plt.savefig("upload_%s_RadarPlot_Merged_cluster.pdf"%today,bbox_inches='tight')
[65]:
scref
[65]:
AnnData object with n_obs × n_vars = 474 × 21397
obs: 'cellID', 'Cluster', 'mtrain_index', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'rank_genes_groups', 'train_set_gene', 'mclasses_names', 'refcolor_dict', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'train_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[70]:
#work_dir=""
#filename="%s_Ref%s_MergeCluster_35epch.h5ad"%("LiverMacrophageNNlearned",today)
[71]:
os.path.join(work_dir,filename)
[71]:
'/Dropbox/data/proj/PE_HYZ/PublicDataSet/Liver_Macrophage/LiverMacrophageNNlearned_Ref2022-12-21_MergeCluster_35epch.h5ad'
[72]:
scm.CamelSwapline.writedata(adatax=scref,filename=filename,filepath=work_dir)
[196]:
scref.obs.to_csv("Metatable.csv",sep=",")