Tutorials_scCAMEL-SWAPLINE_SensoryNeurons_Training-SharmaMouse_Predict_ZeiselMouse
Original Article: Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain,”Nat Commun”, 2021
Original Article: Single-Soma Deep RNA sequencing of Human DRG Neurons Reveals Novel Molecular and Cellular Mechanisms Underlying Somatosensation
Package: scCAMEL-SWAPLINE.v1
Research Group: Ernfors lab
Link of the datasets: Mouse sensory neurons, Dataset references: Sharma, et al., Zeisel, et al.
Training
[1]:
import datetime
today=f"{datetime.datetime.now():%Y-%m-%d}"
today
[1]:
'2023-05-23'
[2]:
import torch
import torch.nn as nn
from torch.autograd import Variable
import torch.utils.data as Data
import torchvision
import matplotlib.pyplot as plt
from mpl_toolkits.mplot3d import Axes3D
import torch.utils.data as data_utils
from matplotlib import cm
import numpy as np
import pandas as pd
import pickle as pickle
from scipy.spatial.distance import cdist, pdist, squareform
import pandas as pd
from sklearn.linear_model import LogisticRegression, LogisticRegressionCV
from sklearn.model_selection import StratifiedShuffleSplit
from collections import defaultdict
from sklearn import preprocessing
import matplotlib.patches as mpatches
import torch.nn.functional as F
import math
#import gpytorch
import urllib.request
import os.path
from scipy.io import loadmat
from math import floor
import anndata
# Make plots inline
%pylab inline
%pylab is deprecated, use %matplotlib inline and import the required libraries.
Populating the interactive namespace from numpy and matplotlib
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/IPython/core/magics/pylab.py:162: UserWarning: pylab import has clobbered these variables: ['floor']
`%matplotlib` prevents importing * from pylab and numpy
warn("pylab import has clobbered these variables: %s" % clobbered +
[3]:
torch.manual_seed(1) # reproducible
[3]:
<torch._C.Generator at 0x7f7df12cde90>
[4]:
import scCAMEL as scm
from scCAMEL import CamelPrefiltering
from scCAMEL import CamelSwapline
from scCAMEL import CamelEvo
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelSwapline.py:637: FutureWarning: The default dtype for empty Series will be 'object' instead of 'float64' in a future version. Specify a dtype explicitly to silence this warning.
def addcolor(datax,clustername="Cluster", colorcode="color", predef=pd.Series()):
[5]:
cd .
/mnt/f/Dropbox/data/proj/PE_HYZ/IpythonRecord/Monkey_DRG
[6]:
cd /mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets
/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets
[7]:
screfall=anndata.read("SensoryNeurons_Zeisel-Sharma.h5ad")
screfall
[7]:
AnnData object with n_obs × n_vars = 12635 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
uns: 'refcolor_dict'
[8]:
#if the matrix is sparse matrix
#screfall.X=screfall.X.todense()
[9]:
set(screfall.obs["Cluster"])
[9]:
{'NF1',
'NF2_3',
'NF4',
'NP1',
'NP2',
'NP3',
'PEP1_PSPEP2',
'PEP1_PSPEP3',
'PEP1_PSPEP4',
'PEP1_PSPEP5',
'PEP2',
'Sharma_Abeta-Field',
'Sharma_Abeta-RA-LTMR',
'Sharma_Adelta-LTMR',
'Sharma_C-LTMR',
'Sharma_CGRP-Alpha',
'Sharma_CGRP-Beta',
'Sharma_CGRP-Epsilion',
'Sharma_CGRP-Eta',
'Sharma_CGRP-Gamma',
'Sharma_CGRP-Theta',
'Sharma_CGRP-Zeta',
'Sharma_Nonpeptidergic nociceptors',
'Sharma_Proprioceptors',
'Sharma_Sst',
'Sharma_TrpM8',
'TRPM8',
'Th'}
[10]:
scref=screfall[screfall.obs["batch"]=="0"]
[11]:
set(scref.obs["Cluster"])
[11]:
{'Sharma_Abeta-Field',
'Sharma_Abeta-RA-LTMR',
'Sharma_Adelta-LTMR',
'Sharma_C-LTMR',
'Sharma_CGRP-Alpha',
'Sharma_CGRP-Beta',
'Sharma_CGRP-Epsilion',
'Sharma_CGRP-Eta',
'Sharma_CGRP-Gamma',
'Sharma_CGRP-Theta',
'Sharma_CGRP-Zeta',
'Sharma_Nonpeptidergic nociceptors',
'Sharma_Proprioceptors',
'Sharma_Sst',
'Sharma_TrpM8'}
[12]:
scref.obs.groupby(["Cluster"]).count()
[12]:
| mtrain_index | sample | species | batch | color | |
|---|---|---|---|---|---|
| Cluster | |||||
| Sharma_Abeta-Field | 257 | 257 | 257 | 257 | 257 |
| Sharma_Abeta-RA-LTMR | 273 | 273 | 273 | 273 | 273 |
| Sharma_Adelta-LTMR | 182 | 182 | 182 | 182 | 182 |
| Sharma_C-LTMR | 1554 | 1554 | 1554 | 1554 | 1554 |
| Sharma_CGRP-Alpha | 1440 | 1440 | 1440 | 1440 | 1440 |
| Sharma_CGRP-Beta | 135 | 135 | 135 | 135 | 135 |
| Sharma_CGRP-Epsilion | 850 | 850 | 850 | 850 | 850 |
| Sharma_CGRP-Eta | 270 | 270 | 270 | 270 | 270 |
| Sharma_CGRP-Gamma | 705 | 705 | 705 | 705 | 705 |
| Sharma_CGRP-Theta | 758 | 758 | 758 | 758 | 758 |
| Sharma_CGRP-Zeta | 333 | 333 | 333 | 333 | 333 |
| Sharma_Nonpeptidergic nociceptors | 2817 | 2817 | 2817 | 2817 | 2817 |
| Sharma_Proprioceptors | 234 | 234 | 234 | 234 | 234 |
| Sharma_Sst | 761 | 761 | 761 | 761 | 761 |
| Sharma_TrpM8 | 488 | 488 | 488 | 488 | 488 |
Prefiltering_and_SelectFeatures
[13]:
path='/mnt/f/Dropbox/data/proj/PE_HYZ/PublicDataSet/'
filename='PANTHER_cell_cycle_genes.txt'
#dfpfc2= prefilteringTest.prefilter(df_f=dfpfc,filename=filename, path=path)
scref= scm.CamelPrefiltering.prefilter(datax=scref,filename=filename, path=path)
CamelRunning_Prefilter......
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelPrefiltering.py:70: ImplicitModificationWarning: Trying to modify attribute `.var` of view, initializing view as actual.
datax.var["Filter1"]=ftlist
CamelRunning_Prefilter......Finished
[14]:
scref=scm.CamelPrefiltering.DataScaling(scref)
[15]:
dfdev2=pd.DataFrame(scref.X,index=scref.obs.index,columns=scref.var.index).T
dfdev2=dfdev2.loc[scref.var['Filter1']==True]
dfdev2.shape
[15]:
(12240, 11057)
[16]:
markerlist=scm.CamelSwapline.enrichmentscoreBETA(dfpfcclus=scref.obs["Cluster"], df_dev=dfdev2, fc=3, pvalcutoff=0.1, shortcut=True)
Camel...Running: clusteringValue1...
Camel...Running: clusteringValue2...
2448
[Processing15
Camel...Running: CrossChecking...
-0%--6%--13%--20%--26%--33%--40%--46%--53%--60%--66%--73%--80%--86%--93%-Camel...Running: output genelist...
[17]:
markerlist2=list(set(markerlist))
len(markerlist2)
[17]:
2738
[18]:
scref, score, thrs, ax=scm.CamelPrefiltering.MVgenes(datax=scref,wt=3, markerlist=markerlist2,
plotfig=True, Xlow=-8.5, Xhigh=6.5, Ylow=-2, Yhigh=6.5,alphaValue=0.9, sValue=30,
fig_args={'figsize': (8, 8), 'facecolor': 'white', 'edgecolor': 'white'})
CamelRunning_MVgenes......
No artists with labels found to put in legend. Note that artists whose label start with an underscore are ignored when legend() is called with no argument.
CamelRunning_MVgenes......Finished
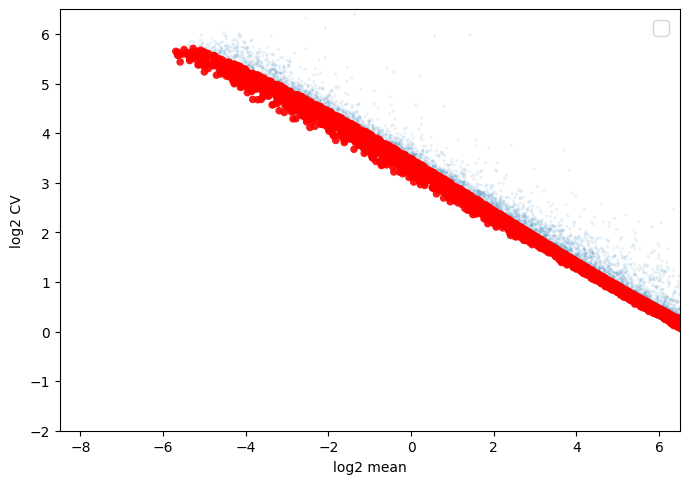
[19]:
len(scref.var.index[scref.var["MVgene"]])
[19]:
5958
[20]:
scref2=scref
[21]:
########################################################
########################################################
#remeber to change the file path in tftable
########################################################
########################################################
scref =scm.CamelPrefiltering.MVgene_Scaling(datax=scref2,score=score,
thrs=thrs, TPTT=100000, mprotogruop=scref.obs["Cluster"].values,commongene=None,
sharedMVgenes=None,std_scaling=True,
tftable="/mnt/f/Dropbox/data/proj/PE_HYZ/PublicDataSet/FantomTF2CLUSTER_human_official.txt", learninggroup="train")
CamelRunning---GenesScaling......
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelPrefiltering.py:791: FutureWarning: In a future version of pandas all arguments of DataFrame.dropna will be keyword-only.
scalepfc = dfpfc.div(dfpfc.std(1), axis=0).dropna(0)
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelPrefiltering.py:792: FutureWarning: In a future version of pandas all arguments of DataFrame.dropna will be keyword-only.
scalepfc = dfpfc.astype(float).dropna(0)
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelPrefiltering.py:794: FutureWarning: In a future version of pandas all arguments of DataFrame.dropna will be keyword-only.
scalepfc = scalepfc.dropna(0)
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelPrefiltering.py:795: FutureWarning: Passing a set as an indexer is deprecated and will raise in a future version. Use a list instead.
dfpfc_dev = scalepfc.loc[set(scalepfc.index) & set(sharedMVgenes)].dropna()
CamelRunning---TrainingGenesScaling......Finished
[22]:
scref
[22]:
AnnData object with n_obs × n_vars = 11057 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names'
obsm: 'train_set_values'
Neural-Network learning
[23]:
net=scm.CamelPrefiltering.NNclassifer(
datax=scref,
epochNum=100,
learningRate=0.0075,
verbose=0,
optimizerMmentum=0.8,
dropout=0.3,
#imizer__nesterov=True,
)
CamelRunning---NNclasffier_in_cpu.......
CamelRunning---NNclasffier_in_cpu.......Finished
Accuracy plot, the overall clustering accuracy is ~85%
[24]:
ax=scm.CamelPrefiltering.AccuracyPlot( nnModel=net, accCutoff=0.8,
Xlow=-1, Ylow=0.0, Yhigh=1,
)
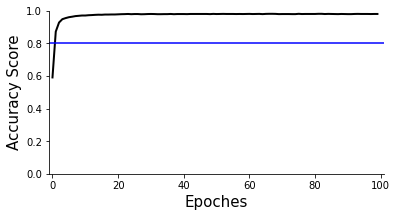
Make predition and visualization in Radar plot
[23]:
net=scm.CamelPrefiltering.NNclassifer(
datax=scref,
epochNum=30,
learningRate=0.0075,
verbose=0,
optimizerMmentum=0.8,
dropout=0.3,
#imizer__nesterov=True,
)
CamelRunning---NNclasffier_in_cpu.......
CamelRunning---NNclasffier_in_cpu.......Finished
[24]:
scref
[24]:
AnnData object with n_obs × n_vars = 11057 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names'
obsm: 'train_set_values'
[25]:
#if color is not defined: scref.obs[ 'color']
#scref=scm.CamelSwapline.addcolor(datax=scref,clustername="Cluster", colorcode="color")
[26]:
scref.uns["mwanted_order"] =list(sort(list(set(scref.obs["Cluster"]))))
[27]:
cd .
/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets
[28]:
#radar plot
scref=scm.CamelSwapline.prediction(datax=scref, mcolor_dict=scref.uns["refcolor_dict"] ,net=net,learninggroup="train", radarplot=True,fontsizeValue=18,
ncolnm=3, bbValue=(1.2, 1.05) )
#plt.savefig("upload_%s_RadarPlot_MacaqueRef_Merged_cluster.pdf"%today,bbox_inches='tight')

[29]:
scref
[29]:
AnnData object with n_obs × n_vars = 11057 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'train_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
dfpfcclus = scref.obs[[“mtrain_index”,”Cluster”]].T dfpfcclus
[ ]:
#######################
#######################
#Multiplot_Violin
# each column along X axis represents a cell type, and the y-axis represents the cell-type probability
# each column along X axis contain all cells in the training, shown as dots with different colors
# in the perfect condition, only the cells of the same cell-type column will reach to the maxium (100%) probability, all other cells will be 0 probability
#however, in each cell-type column, some cells from other cell types may exhibit high probability, two reasons: 1. they may shared some features with this---
# cell-type, and these features drag them to the current cell-type; 2. they are inproperly classified due to many factors, such as low quality data, different clustering algorithm.....
#######################
#######################
[31]:
fig=scm.CamelSwapline.ProbMultiPlot( datax=scref, mcolor_dict=scref.uns["refcolor_dict"] )
<Figure size 2500x1000 with 0 Axes>
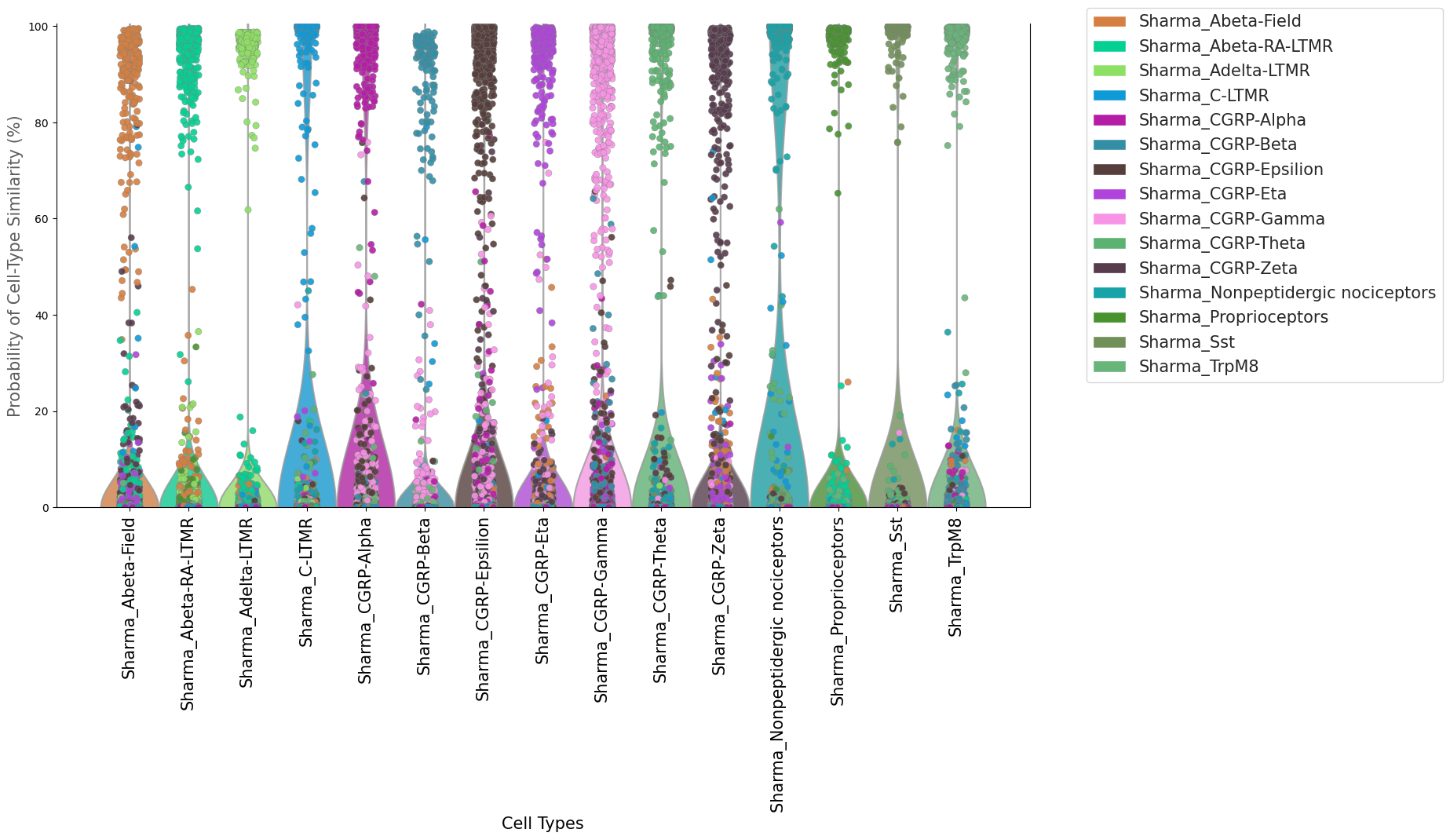
permutation control
[32]:
## the whole data matrix is randomized, the red X represents 95% conficence of each cell-type
[33]:
dftest0, ratiodf=scm.CamelSwapline.permutationTest(datax=scref,net=net,num=50, plotshow=True)
<Figure size 640x480 with 0 Axes>
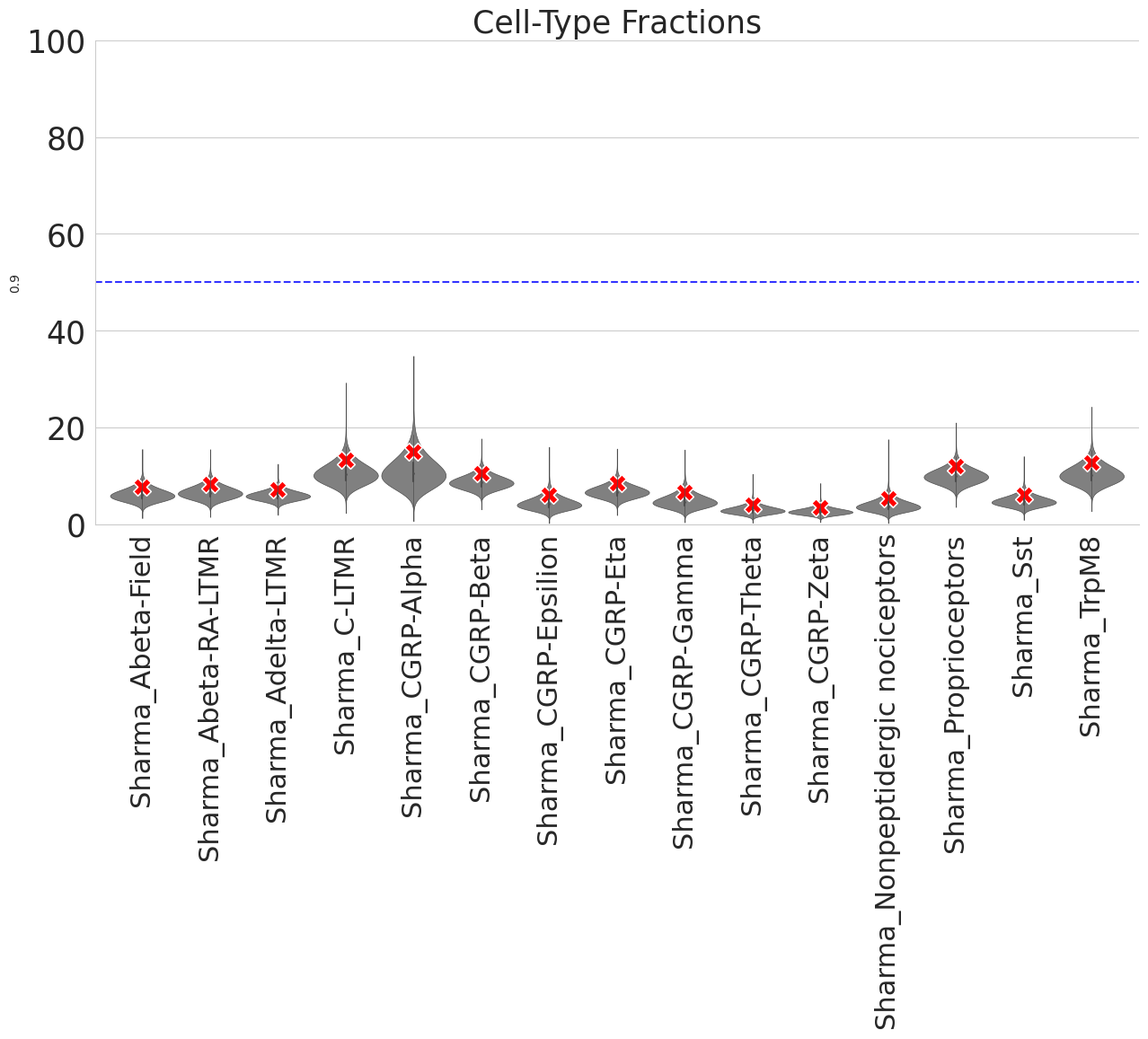
Cluster consistency and accuracy
[ ]:
######
# The script uses the original, defined cell type as a reference to compare with the predicted cell type of each cell.
# If the predicted cell type matches the original defined cell type, the cell is deemed to be 'consistently assigned'.
# Conversely, if the predicted cell type does not match, the cell is considered 'inconsistently assigned', indicating possible incorrect clustering.
# Output File: This contains the ID of inconsistent cells, the originally defined cell type, and the predicted cell type.
# Output Plot: This displays the ratio (%) of cells that are correctly (consistently) and incorrectly (inconsistently) assigned cell types.
######
[35]:
len(set(scref.obs.index))
[35]:
11057
[36]:
scref
[36]:
AnnData object with n_obs × n_vars = 11057 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'train_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[37]:
cd .
/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets
[38]:
scref=scm.CamelSwapline.ConsistantAssign(datax=scref,dfsig=dftest0,outputfilepath="upload_%s_Sharma_Ref_InconsistantCells.csv"%today,
outputPlot=True )
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelSwapline.py:466: FutureWarning: The frame.append method is deprecated and will be removed from pandas in a future version. Use pandas.concat instead.
dfprob1=dfprob1posi.append(dfprob1neg)
<Figure size 2500x1000 with 0 Axes>
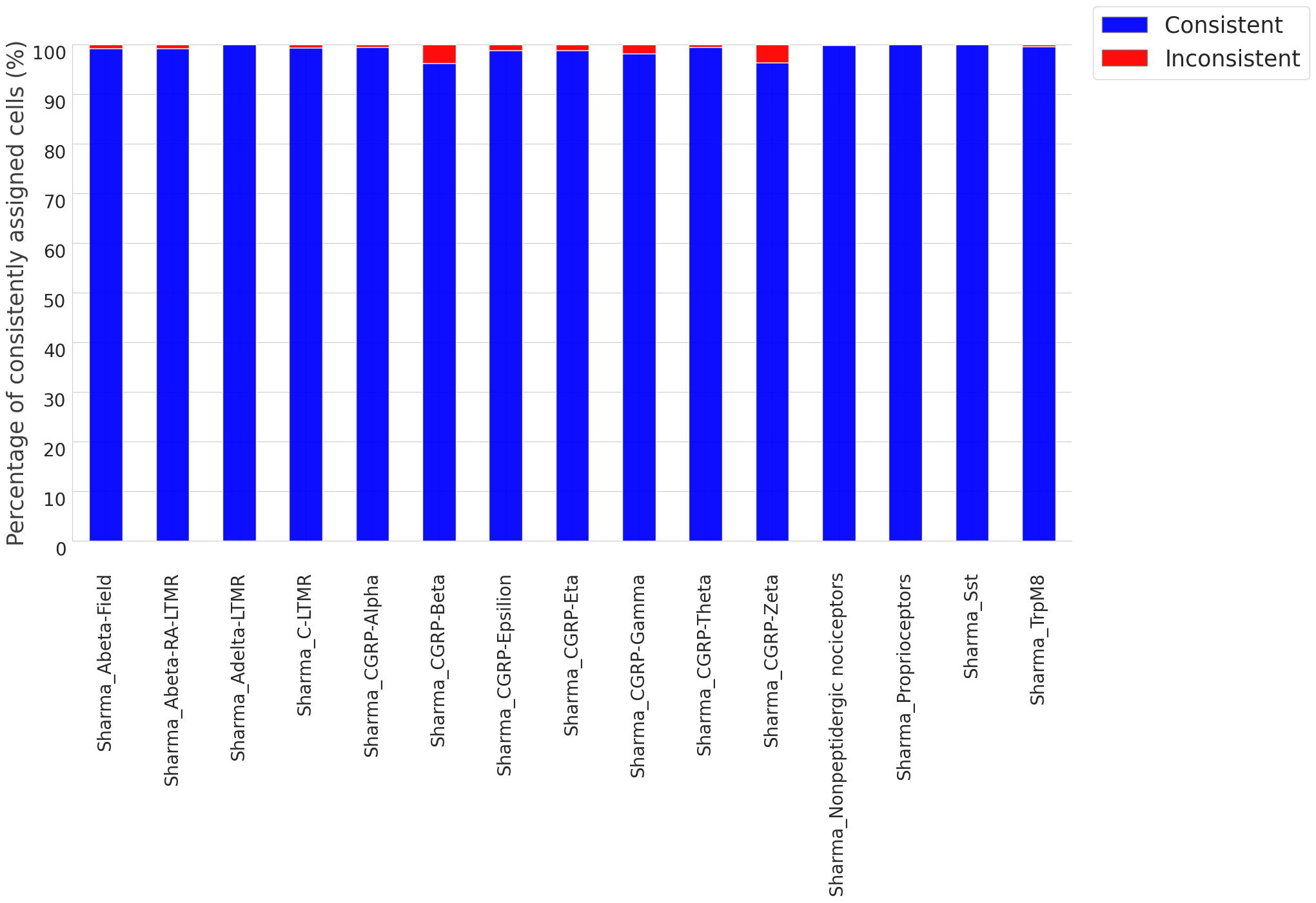
Cell_Type Purity
[39]:
#The ratio of the purity entropy for each cluster based on their learning scores, is used as a measure of purity.
#The function returns a pandas dataframe sorted by the purity score
[40]:
dfpurity1=scm.CamelSwapline.PurityEstimationLearningScore(datax=scref, clusterlist="Cluster", elbow=False, figureplot=True)
<Figure size 640x480 with 0 Axes>
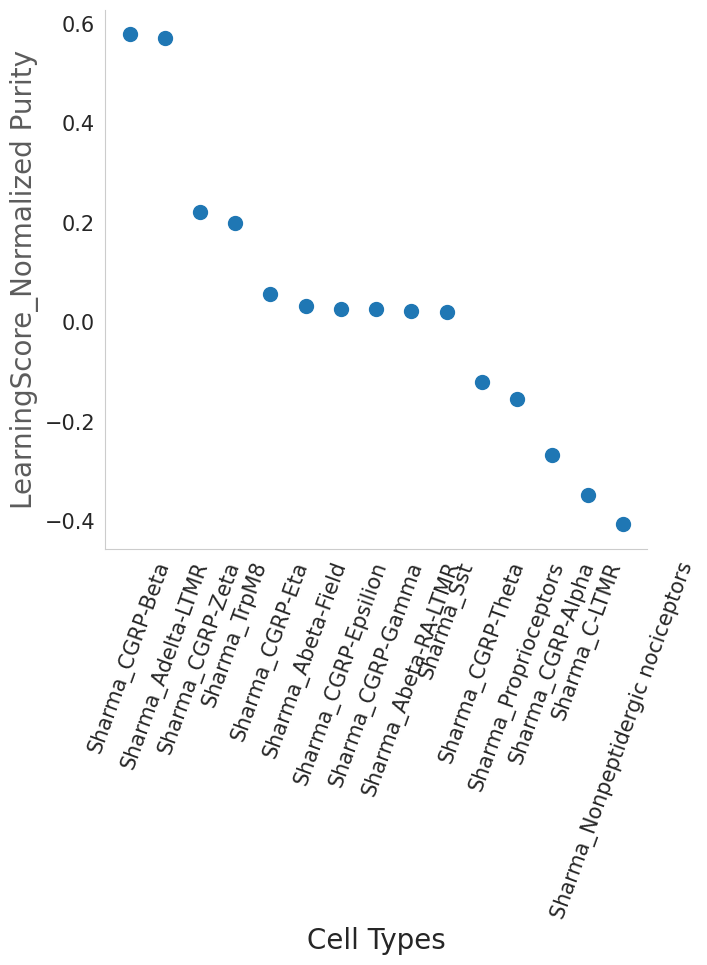
association between cell-types
[41]:
scref
[41]:
AnnData object with n_obs × n_vars = 11057 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color', 'PredictCluster', 'ClusterConsistanceScore'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'train_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[42]:
# the heatmap of hierarchical clustering represents the cell-type similarity or association
#color from dark purple to light yellow represents the association from low to high
#number inside of eahc square indicating the association value.
[43]:
scm.CamelSwapline.CellTypeSimilarity(datax=scref, labelnum=True, metricvalue='correlation',methodvalue="complete")
<Figure size 1500x1500 with 0 Axes>

[44]:
scm.CamelSwapline.CellTypeSimilarity(datax=scref, labelnum=False, metricvalue='correlation',methodvalue="complete")
<Figure size 1500x1500 with 0 Axes>
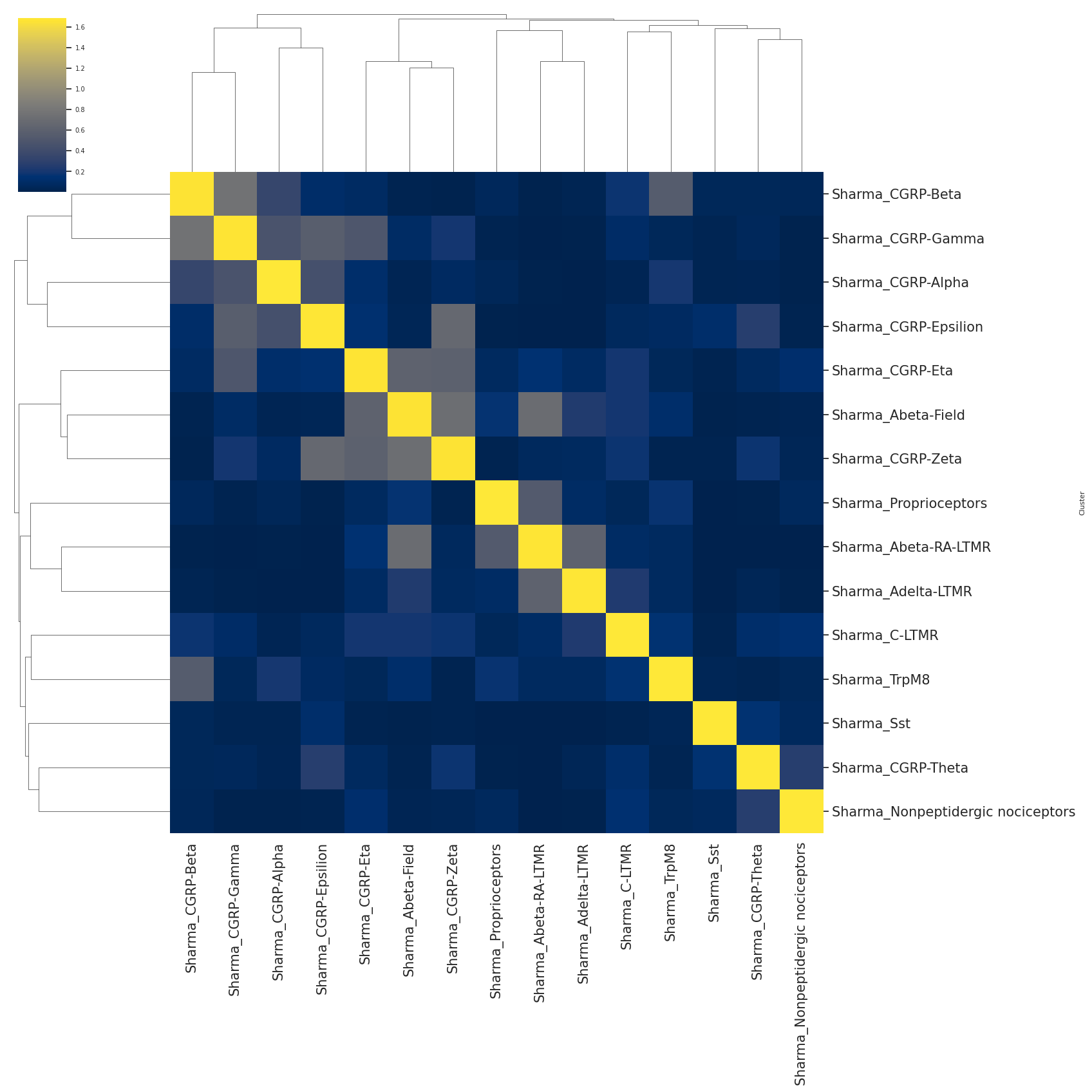
[ ]:
Save data
[45]:
scref
[45]:
AnnData object with n_obs × n_vars = 11057 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color', 'PredictCluster', 'ClusterConsistanceScore'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'train_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[46]:
cd .
/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets
[47]:
work_dir="/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets"
QueryName="SharmaMouse"
TrainingName="SharmaMouse"
filename="%s_%s_Ref%s_MergeCluster.h5ad"%(QueryName,TrainingName,today)
[48]:
os.path.join(work_dir,filename)
[48]:
'/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets/SharmaMouse_SharmaMouse_Ref2023-05-23_MergeCluster.h5ad'
[49]:
CamelSwapline.writedata(adatax=scref,filename=filename,filepath=work_dir)
[50]:
scref.X=scref.X.todense()
[ ]:
Prediction
ZeiselMouse_cluster
[51]:
scpdt=screfall[screfall.obs["batch"]=="1"]
[52]:
set(scpdt.obs["Cluster"])
[52]:
{'NF1',
'NF2_3',
'NF4',
'NP1',
'NP2',
'NP3',
'PEP1_PSPEP2',
'PEP1_PSPEP3',
'PEP1_PSPEP4',
'PEP1_PSPEP5',
'PEP2',
'TRPM8',
'Th'}
[53]:
scpdt
[53]:
View of AnnData object with n_obs × n_vars = 1578 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
uns: 'refcolor_dict'
[54]:
scpdt.var.index
[54]:
Index(['HTR1F', 'GPAT2', 'CMTM7', 'SST', 'NTS', 'IL31RA', 'NPPB', 'ADA',
'CFAP61', 'CBLN1',
...
'KRTAP7-1', 'NKX1-1', 'NKX1-2', 'NKX2-2', 'NKX3-1', 'NKX3-2', 'NKX6-1',
'NKX6-2', 'NKX6-3', 'RPL9'],
dtype='object', length=15677)
[55]:
scpdt2=scpdt.copy()
scpdt2.X=scpdt.X*(scref.X.std()/scpdt2.X.std())
[56]:
########################################################
########################################################
#remeber to change the file path in tftable
########################################################
########################################################
scpdt =scm.CamelPrefiltering.MVgene_Scaling(datax=scpdt2,TPTT=1000000, commongene=scref.var.index.tolist(),
sharedMVgenes=scref.uns[ 'train_set_gene'].tolist(),
std_scaling=True,score=None, thrs=None, mprotogruop=None,
tftable="f:/Dropbox/data/proj/PE_HYZ/PublicDataSet/FantomTF2CLUSTER_human_official.txt", learninggroup="test")
CamelRunning---GenesScaling......
/home/huyiz/anaconda3/envs/newpy38/lib/python3.8/site-packages/scCAMEL/CamelPrefiltering.py:820: FutureWarning: In a future version of pandas all arguments of DataFrame.dropna will be keyword-only.
scalegbm = dfpfc.div(dfpfc.std(1), axis=0).dropna(0)
CamelRunning---TestGenesScaling......Finished
[57]:
scpdt
[57]:
AnnData object with n_obs × n_vars = 1578 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names'
obsm: 'test_set_values'
[58]:
#del scpdt.obs["color"]
[59]:
# if color is not definedi
#scpdt=scm.CamelSwapline.addcolor(datax=scpdt,clustername="Cluster", colorcode="color")
[60]:
scpdt.uns["mwanted_order"] =list(sort(list(set(scpdt.obs["Cluster"]))))
[61]:
scpdt
[61]:
AnnData object with n_obs × n_vars = 1578 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names', 'mwanted_order'
obsm: 'test_set_values'
[62]:
test=scm.CamelSwapline.prediction(datax=scpdt, mcolor_dict=scpdt.uns["refcolor_dict"],net=net,learninggroup="test", radarplot=True, fontsizeValue=35,
datarefplot=scref, ncolnm=1, bbValue=(1.1, 1.05))

[63]:
scpdt
[63]:
AnnData object with n_obs × n_vars = 1578 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'test_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[64]:
genename=sort(list(set(scpdt.obs["Cluster"])))
name=sort(list(set(scref.obs["Cluster"])))
[65]:
dfprob=pd.DataFrame(scpdt.obsm['Celltype_Score'])
dfprob.columns=scpdt.uns['Celltype_Score_RefCellType']
dfprob.index=scpdt.obs.index
dfmk=dfprob.astype(float).join(scpdt.obs["Cluster"],how="inner").T
dfprob=scm.CamelSwapline.CellTypeSimilarityViolinPlot(datax=scpdt, dataref=scref)
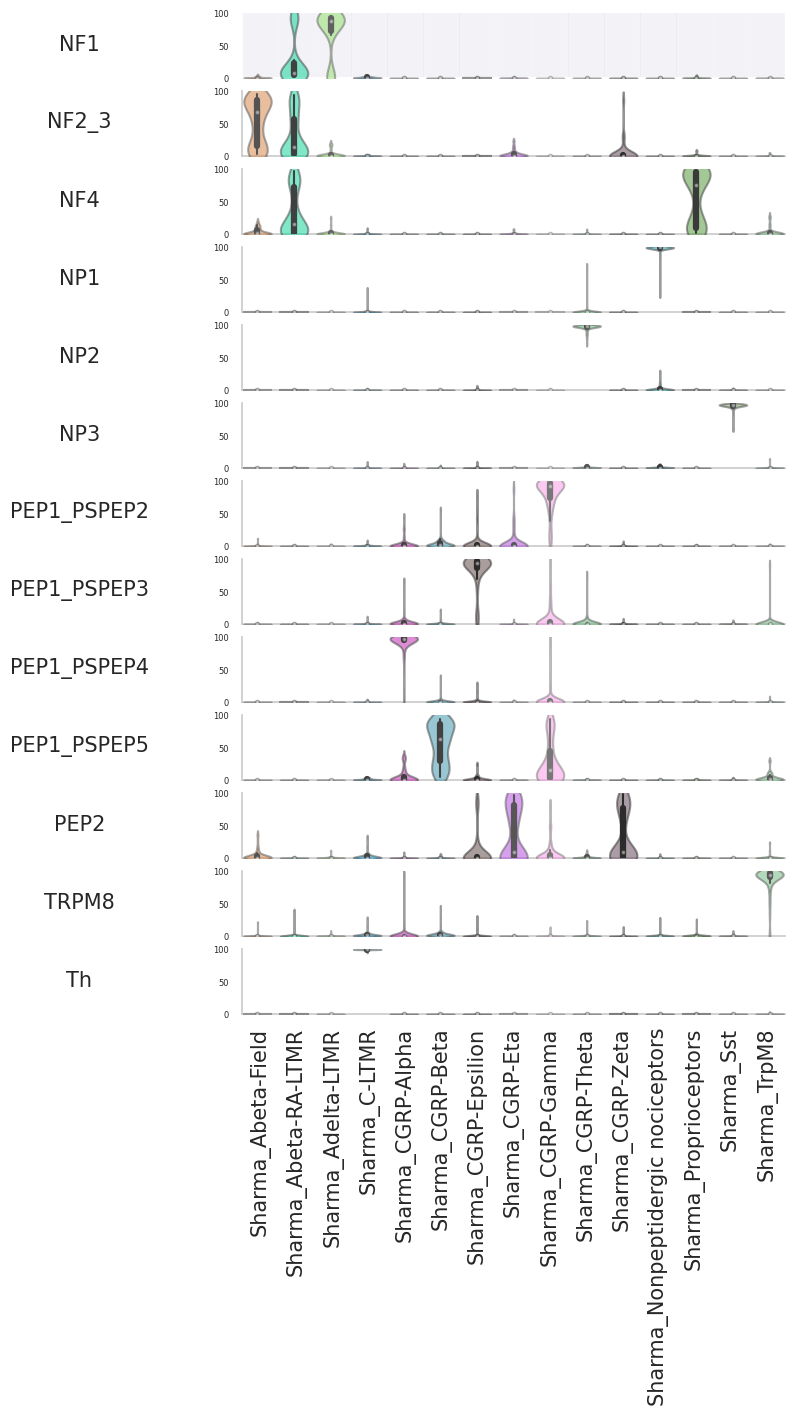
Save data
[66]:
cd .
/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets
[67]:
scpdt
[67]:
AnnData object with n_obs × n_vars = 1578 × 15677
obs: 'Cluster', 'mtrain_index', 'sample', 'species', 'batch', 'color'
var: 'RefGeneList'
uns: 'refcolor_dict', 'train_set_gene', 'mclasses_names', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'test_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[68]:
work_dir="/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets"
QueryName="ZeiselMouse"
TrainingName="SharmaMouse"
filename="%s_%s_Ref%s_MergeCluster.h5ad"%(QueryName,TrainingName,today)
[69]:
os.path.join(work_dir,filename)
[69]:
'/mnt/f/Yizhou_KI_OneCloud/OneDrive - Karolinska Institutet/Datasets_Template/Datasets/ZeiselMouse_SharmaMouse_Ref2023-05-23_MergeCluster.h5ad'
[70]:
CamelSwapline.writedata(adatax=scref,filename=filename,filepath=work_dir)
[ ]: