Tutorial: scCAMEL-SWAPLINE Mouse Dentate Gyrus to Human Glioblastoma
This tutorial demonstrates the SWAPLINE workflow with the published PyPI scCAMEL package. It trains a reference neural-network classifier on mouse dentate gyrus clusters, saves/reloads the model, and predicts human glioblastoma lineage-like cluster signals.
Original Article: Neural network learning defines glioblastoma features to be of neural crest perivascular or radial glia lineages,”Sci. Adv.”, 2022
Package: scCAMEL from PyPI, tested here with version 0.47b0.
Method: scCAMEL-SWAPLINE.v1
Author: Yizhou Hu, Research Group: Ernfors lab
Link of the datasets: Mouse Dentate Gyrus, Human glioblastoma, Dataset references: Hochgerner and Zeisel, et al., Couturier, et al.
Resource gene listcell cycle genes, Homologene-HumanMouse
Training
[1]:
import datetime
today=f"{datetime.datetime.now():%Y-%m-%d}"
today
[1]:
'2026-06-09'
[2]:
import torch
import torch.nn as nn
from torch.autograd import Variable
import torch.utils.data as Data
import torchvision
import matplotlib.pyplot as plt
from mpl_toolkits.mplot3d import Axes3D
import torch.utils.data as data_utils
from matplotlib import cm
import numpy as np
import pandas as pd
import pickle as pickle
from scipy.spatial.distance import cdist, pdist, squareform
import pandas as pd
from sklearn.linear_model import LogisticRegression, LogisticRegressionCV
from sklearn.model_selection import StratifiedShuffleSplit
from collections import defaultdict
from sklearn import preprocessing
import matplotlib.patches as mpatches
import torch.nn.functional as F
import math
#import gpytorch
import urllib.request
import os.path
import os
import sys
import importlib.metadata as importlib_metadata
from pathlib import Path
from scipy.io import loadmat
from math import floor
import anndata
PROJECT_ROOT = Path("/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse")
PUBLIC_DATASET = Path("/mnt/f/Dropbox/data/proj/PE_HYZ/PublicDataSet")
VERSION_ROOT = Path("/mnt/e/Loal_Temp/Vicuna_Example/scCAMEL_VICUNA_updated_20260605")
OUTPUT_DIR = VERSION_ROOT / "outputs" / "swapline_pypi047b0_tutorial"
OUTPUT_DIR.mkdir(parents=True, exist_ok=True)
os.chdir(PROJECT_ROOT)
# Make plots inline
%pylab inline
%pylab is deprecated, use %matplotlib inline and import the required libraries.
Populating the interactive namespace from numpy and matplotlib
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/IPython/core/magics/pylab.py:162: UserWarning: pylab import has clobbered these variables: ['floor']
`%matplotlib` prevents importing * from pylab and numpy
warn("pylab import has clobbered these variables: %s" % clobbered +
[3]:
torch.manual_seed(1) # reproducible
[3]:
<torch._C.Generator at 0x7b5b057d6410>
[4]:
import scCAMEL as scm
from scCAMEL import CamelPrefiltering
from scCAMEL import CamelSwapline
from scCAMEL import CamelEvo
SC_CAMEL_VERSION = importlib_metadata.version("scCAMEL")
print("Using installed scCAMEL:", SC_CAMEL_VERSION)
print("scCAMEL import path:", Path(scm.__file__).parent)
print("Tutorial outputs:", OUTPUT_DIR)
Using installed scCAMEL: 0.47b0
scCAMEL import path: /home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL
Tutorial outputs: /mnt/e/Loal_Temp/Vicuna_Example/scCAMEL_VICUNA_updated_20260605/outputs/swapline_pypi047b0_tutorial
[5]:
os.chdir(PROJECT_ROOT)
Path.cwd()
[5]:
PosixPath('/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse')
[6]:
scref=anndata.read(PROJECT_ROOT / "ZeiselDentateGyrus_Ref2023-05-27.h5ad")
scref
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/__init__.py:42: FutureWarning: `anndata.read` is deprecated, use `anndata.read_h5ad` instead. `ad.read` will be removed in mid 2024.
warnings.warn(
[6]:
AnnData object with n_obs × n_vars = 5454 × 14545
obs: 'Cluster', 'Color'
[7]:
set(scref.obs["Cluster"])
[7]:
{'Astrocytes',
'Cajal-Retzius',
'Cck-Tox',
'Endo',
'GABA',
'Granule',
'Microglia',
'Mossy',
'NFOL',
'Neuroblast',
'OLIG',
'OPC',
'PVM',
'Peri/VLMC',
'nIPC/Rgl'}
[8]:
scref.obs.groupby(["Cluster"]).count()
/tmp/ipykernel_44346/1119954423.py:1: FutureWarning: The default of observed=False is deprecated and will be changed to True in a future version of pandas. Pass observed=False to retain current behavior or observed=True to adopt the future default and silence this warning.
scref.obs.groupby(["Cluster"]).count()
[8]:
| Color | |
|---|---|
| Cluster | |
| Astrocytes | 335 |
| Cajal-Retzius | 93 |
| Cck-Tox | 27 |
| Endo | 165 |
| GABA | 99 |
| Granule | 3045 |
| Microglia | 169 |
| Mossy | 137 |
| NFOL | 35 |
| Neuroblast | 874 |
| OLIG | 79 |
| OPC | 66 |
| PVM | 18 |
| Peri/VLMC | 59 |
| nIPC/Rgl | 253 |
[9]:
#if the matrix is sparse matrix
#screfall.X=screfall.X.todense()
[10]:
# Apply the function to balance clusters and label up-sampled items
scref= scm.CamelPrefiltering.balance_clusters_adata(scref, 'Cluster',5)
scref
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/_core/anndata.py:1756: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
[10]:
AnnData object with n_obs × n_vars = 13371 × 14545
obs: 'Cluster', 'Color', 'upsampled'
[11]:
scref.obs.groupby(["Cluster"]).count()
/tmp/ipykernel_44346/1119954423.py:1: FutureWarning: The default of observed=False is deprecated and will be changed to True in a future version of pandas. Pass observed=False to retain current behavior or observed=True to adopt the future default and silence this warning.
scref.obs.groupby(["Cluster"]).count()
[11]:
| Color | upsampled | |
|---|---|---|
| Cluster | ||
| Astrocytes | 944 | 944 |
| Cajal-Retzius | 702 | 702 |
| Cck-Tox | 636 | 636 |
| Endo | 774 | 774 |
| GABA | 708 | 708 |
| Granule | 3045 | 3045 |
| Microglia | 778 | 778 |
| Mossy | 746 | 746 |
| NFOL | 644 | 644 |
| Neuroblast | 874 | 874 |
| OLIG | 688 | 688 |
| OPC | 675 | 675 |
| PVM | 627 | 627 |
| Peri/VLMC | 668 | 668 |
| nIPC/Rgl | 862 | 862 |
Prefiltering_and_SelectFeatures
[12]:
# if needed scref.X=scref.X.todense()
scref.X=scref.X.todense()
scref=scm.CamelPrefiltering.DataScaling(scref)
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/_core/storage.py:39: ImplicitModificationWarning: X should not be a np.matrix, use np.ndarray instead.
warnings.warn(msg, ImplicitModificationWarning)
[13]:
dfpdt=pd.DataFrame(scref.X.T,index=scref.var.index,columns=scref.obs.index)
dfpdt.shape
[13]:
(14545, 13371)
[14]:
path=str(PUBLIC_DATASET) + "/"
dictfilename1="Homologene_mouse2human_dict2.pickle"
dfpdt= scm.CamelPrefiltering.TransSpeciesGeneName(dfm=dfpdt, dictfilename=dictfilename1, path=path)
samegene=set(dfpdt.index)
len(samegene)
[14]:
12516
[15]:
dfpdt
[15]:
| 10X46_1_ACTCTATGGTACGT-1 | 10X43_1_AGGGACGATCTCCG-1 | 10X46_1_GACAACTGTTGACG-1 | 10X46_1_CTGTAACTGGTCTA-1 | 10X43_1_TGCAAGTGTCTCCG-1 | 10X43_1_GTAACGTGCCTAAG-1 | 10X43_1_ATCACGGAGGAGTG-1 | 10X43_1_AATTGTGAGTTGCA-1 | 10X43_1_TGGTCAGACTATTC-1 | 10X43_1_ACAGTCGAGGGACA-1 | ... | 10X43_1_CTTTCAGAAACCAC-1 | 10X43_1_ATAACCCTCCTCAC-1 | 10X43_1_GCAGGCACACCCAA-1 | 10X43_1_CTTTCAGAAACCAC-1 | 10X43_1_CAACGAACCAGTCA-1 | 10X46_1_TGAACCGAATGCCA-1 | 10X43_1_GCAGGCACACCCAA-1 | 10X46_1_CTCGCATGTATCTC-1 | 10X43_1_GTATTCACCTAGTG-1 | 10X46_1_AAGACAGACAGTTG-1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A2M | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| AAAS | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| AACS | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| AAED1 | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.57404 | 0.000000 | 0.0 | 0.57404 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| AAGAB | 0.0 | 0.000000 | 0.700669 | 0.826938 | 0.000000 | 1.424036 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 1.042694 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.0 | 1.374609 | 0.0 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| ZXDC | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| ZYG11B | 0.0 | 0.000000 | 1.401339 | 0.826938 | 1.417607 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| ZYX | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.708804 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 1.042694 | 0.0 | 0.00000 | 1.837025 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| ZZEF1 | 0.0 | 0.696231 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.57404 | 0.000000 | 0.0 | 0.57404 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
| ZZZ3 | 0.0 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.0 | ... | 0.00000 | 0.000000 | 0.0 | 0.00000 | 0.000000 | 0.0 | 0.0 | 0.0 | 0.000000 | 0.0 |
12516 rows × 13371 columns
[16]:
scref2= anndata.AnnData(dfpdt.T.astype(float))
scref2
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/_core/anndata.py:1756: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
[16]:
AnnData object with n_obs × n_vars = 13371 × 12516
[17]:
scref2.obs=scref.obs
[18]:
scref=scref2.copy()
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/_core/anndata.py:1756: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
[19]:
path=str(PUBLIC_DATASET) + '/'
filename='PANTHER_cell_cycle_genes.txt'
scref= scm.CamelPrefiltering.prefilter(datax=scref,filename=filename, path=path)
CamelRunning_Prefilter......
CamelRunning_Prefilter......Finished
[20]:
scref=scm.CamelPrefiltering.DataScaling(scref)
[21]:
scref.obs
[21]:
| Cluster | Color | upsampled | |
|---|---|---|---|
| 10X46_1_ACTCTATGGTACGT-1 | Granule | #b48c82 | False |
| 10X43_1_AGGGACGATCTCCG-1 | Granule | #b48c82 | False |
| 10X46_1_GACAACTGTTGACG-1 | Granule | #b48c82 | False |
| 10X46_1_CTGTAACTGGTCTA-1 | Granule | #b48c82 | False |
| 10X43_1_TGCAAGTGTCTCCG-1 | Granule | #b48c82 | False |
| ... | ... | ... | ... |
| 10X46_1_TGAACCGAATGCCA-1 | PVM | #8b6564 | True |
| 10X43_1_GCAGGCACACCCAA-1 | PVM | #8b6564 | True |
| 10X46_1_CTCGCATGTATCTC-1 | PVM | #8b6564 | True |
| 10X43_1_GTATTCACCTAGTG-1 | PVM | #8b6564 | True |
| 10X46_1_AAGACAGACAGTTG-1 | PVM | #8b6564 | True |
13371 rows × 3 columns
[22]:
scref=scm.CamelPrefiltering.SelectFeatures(datax=scref, clustername='Cluster',methodname='Enrichment_shortcut', numbergenes=50, folderchange=1.5)
Camel...Running: clusteringValue1...
Camel...Running: clusteringValue2...
[Processing]-0%--6%--13%--20%--26%--33%--40%--46%--53%--60%--66%--73%--80%--86%--93%-Camel...Running: CrossChecking...
Camel...Running: output genelist...
[23]:
len(scref.var.index[scref.var["MVgene"]])
[23]:
2246
[24]:
scref2=scref.copy()
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/_core/anndata.py:1756: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
[25]:
########################################################
########################################################
#remeber to change the file path in tftable
########################################################
scref =scm.CamelPrefiltering.LabelGene_Scaling(datax=scref2,
TPTT=100000, mprotogruop=scref2.obs["Cluster"].values, commongene=None,
sharedMVgenes=None, std_scaling=True,
tftable=str(PUBLIC_DATASET / "FantomTF2CLUSTER_human_official.txt"), learninggroup="train")
CamelRunning---GenesScaling......
CamelRunning---TrainingGenesScaling......Finished
[26]:
scref
[26]:
AnnData object with n_obs × n_vars = 13371 × 12516
obs: 'Cluster', 'Color', 'upsampled', 'mtrain_index'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'train_set_gene', 'mclasses_names'
obsm: 'train_set_values'
Neural-Network learning
[27]:
net=scm.CamelPrefiltering.NNclassifer(
datax=scref,
epochNum=150,
learningRate=0.005,
verbose=0,
optimizerMmentum=0.8,
dropout=0.3,
#imizer__nesterov=True,
)
CamelRunning---NNclasffier_in_cpu.......
CamelRunning---NNclasffier_in_cpu.......Finished
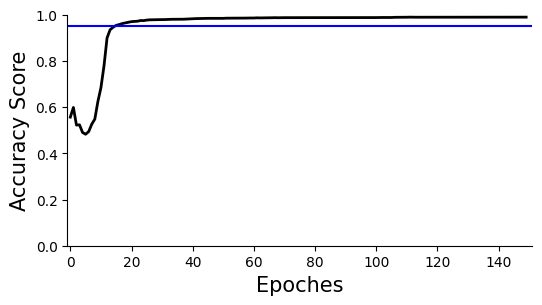
Accuracy plot, the overall clustering accuracy is ~95%
[28]:
ax=scm.CamelPrefiltering.AccuracyPlot( nnModel=net, accCutoff=0.95,
Xlow=-1, Ylow=0.0, Yhigh=1,
)
Save and Reload the Trained Model
Note: The published
scCAMEL 0.47b0package includesCamelSwapline.save_camel_modelandCamelSwapline.load_camel_model, so the tutorial uses those package helpers instead of redefining model serialization inside the notebook.
[29]:
Path.cwd()
[29]:
PosixPath('/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse')
Tutorial note: The checkpoint files are written to
OUTPUT_DIR / "camel_checkpoints2"so they stay with the rest of the tutorial outputs.
[30]:
Path.cwd()
[30]:
PosixPath('/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse')
[31]:
# after net.fit(...)
paths = scm.CamelSwapline.save_camel_model(scref, net, out_dir=str(OUTPUT_DIR / "camel_checkpoints2"), prefix="camel_nn_mouseDG")
print("Saved files:", paths)
Saved files: {'meta': '/mnt/e/Loal_Temp/Vicuna_Example/scCAMEL_VICUNA_updated_20260605/outputs/swapline_pypi047b0_tutorial/camel_checkpoints2/camel_nn_mouseDG_meta.json', 'weights': '/mnt/e/Loal_Temp/Vicuna_Example/scCAMEL_VICUNA_updated_20260605/outputs/swapline_pypi047b0_tutorial/camel_checkpoints2/camel_nn_mouseDG_weights.pt', 'history': '/mnt/e/Loal_Temp/Vicuna_Example/scCAMEL_VICUNA_updated_20260605/outputs/swapline_pypi047b0_tutorial/camel_checkpoints2/camel_nn_mouseDG_history.json'}
[32]:
net3= scm.CamelSwapline.load_camel_model(checkpoint_dir=str(OUTPUT_DIR / "camel_checkpoints2"), prefix="camel_nn_mouseDG",dropoutVal=0.3, device="cpu")
[52]:
[33]:
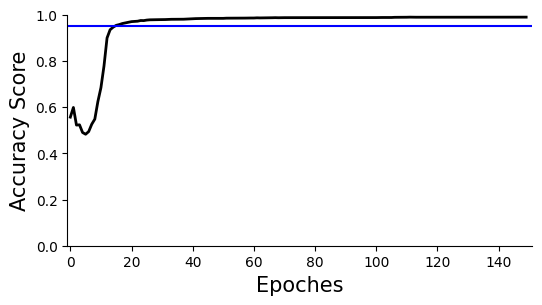
ax=scm.CamelPrefiltering.AccuracyPlot( nnModel=net3, accCutoff=0.95,
Xlow=-1, Ylow=0.0, Yhigh=1,
)
[ ]:
Make predition and visualization in Radar plot
[34]:
net=scm.CamelPrefiltering.NNclassifer(
datax=scref,
epochNum=40,
learningRate=0.005,
verbose=0,
optimizerMmentum=0.8,
dropout=0.3,
#imizer__nesterov=True,
)
CamelRunning---NNclasffier_in_cpu.......
CamelRunning---NNclasffier_in_cpu.......Finished
[35]:
scref
[35]:
AnnData object with n_obs × n_vars = 13371 × 12516
obs: 'Cluster', 'Color', 'upsampled', 'mtrain_index'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'train_set_gene', 'mclasses_names'
obsm: 'train_set_values'
[36]:
#if color is not defined: scref.obs[ 'color']
predefined_colors = pd.Series({
'Astrocytes': [190, 10, 10],'Cajal-Retzius': [225, 160, 30],'Cck-Tox': [217, 215, 7],
'Endo': [170, 180, 170], 'GABA': [130, 140, 140],'Granule': [180, 140, 130],
'Microglia': [100, 100, 240],'Mossy': [ 80, 235, 255],'NFOL':[190, 235, 255],
'Neuroblast':[210, 255, 215],'OLIG':[230, 140, 120], 'OPC': [255, 195, 28],
'PVM': [139, 101, 100],'Pericytes': [252, 183, 26],'Radial Glia-like': [214, 194, 39],
'VLMC': [255, 120, 155],'nIPC': [250, 145, 45],'hRgl2a': [250, 125, 25],
'hDA0': [190, 200, 190],'hOPC': [255, 35, 155],'hRN': [199, 121, 41],
'hNbGaba': [ 40, 55, 130],'hGaba': [ 7, 121, 61],'hOMTN': [ 95, 186, 70],
'hSert': [ 50, 180, 180],'nIPC/Rgl': [245, 205, 170], 'Peri/VLMC': [185, 245, 30],
'eSCc':[205,205,220]
})
[37]:
#if color is not defined
#del scref.obs["color"]
#scref=scm.CamelSwapline.addcolor(datax=scref,clustername="Cluster", colorcode="color")
scref = scm.CamelSwapline.add_color2(scref, clustername="Cluster", colorcode="color",predef=predefined_colors)
[38]:
#if want to define your own order
#scref.uns["mwanted_order"] =[ 'Mossy', 'Cajal-Retzius', 'Cck-Tox', 'GABA', 'Endo', 'Peri/VLMC', 'PVM', 'Microglia', 'Astrocytes', 'OLIG',
#'NFOL', 'OPC', 'nIPC/Rgl','Neuroblast','Granule']
scref.uns["mwanted_order"] =list(sort(list(set(scref.obs["Cluster"]))))
[39]:
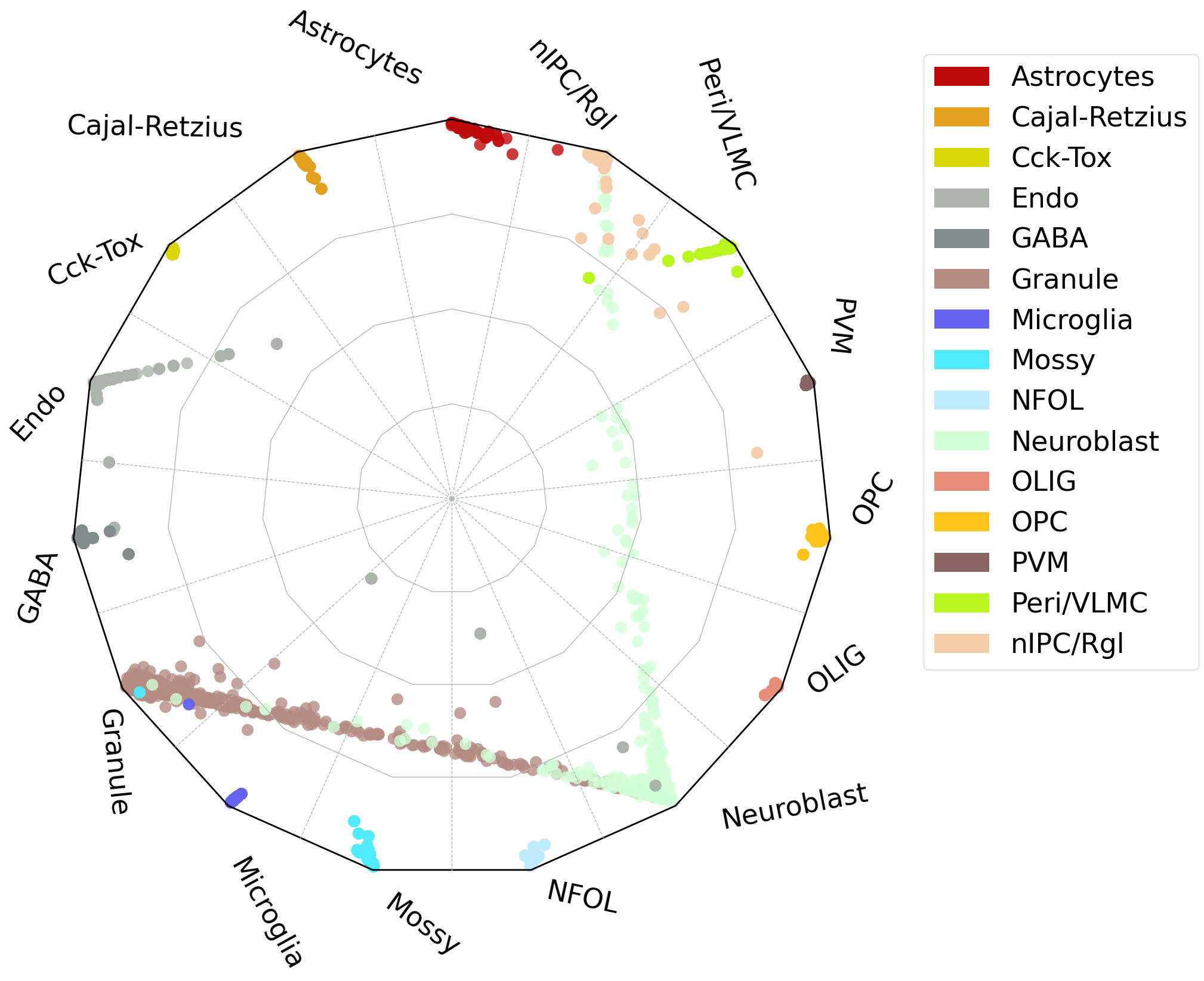
#radar plot
scref=scm.CamelSwapline.prediction(datax=scref, mcolor_dict=scref.uns["refcolor_dict"] ,net=net,learninggroup="train", radarplot=True,fontsizeValue=10,
ncolnm=3, bbValue=(1.5, 1.55) )
#plt.savefig("upload_%s_RadarPlot_cluster.pdf"%today,bbox_inches='tight')
[40]:
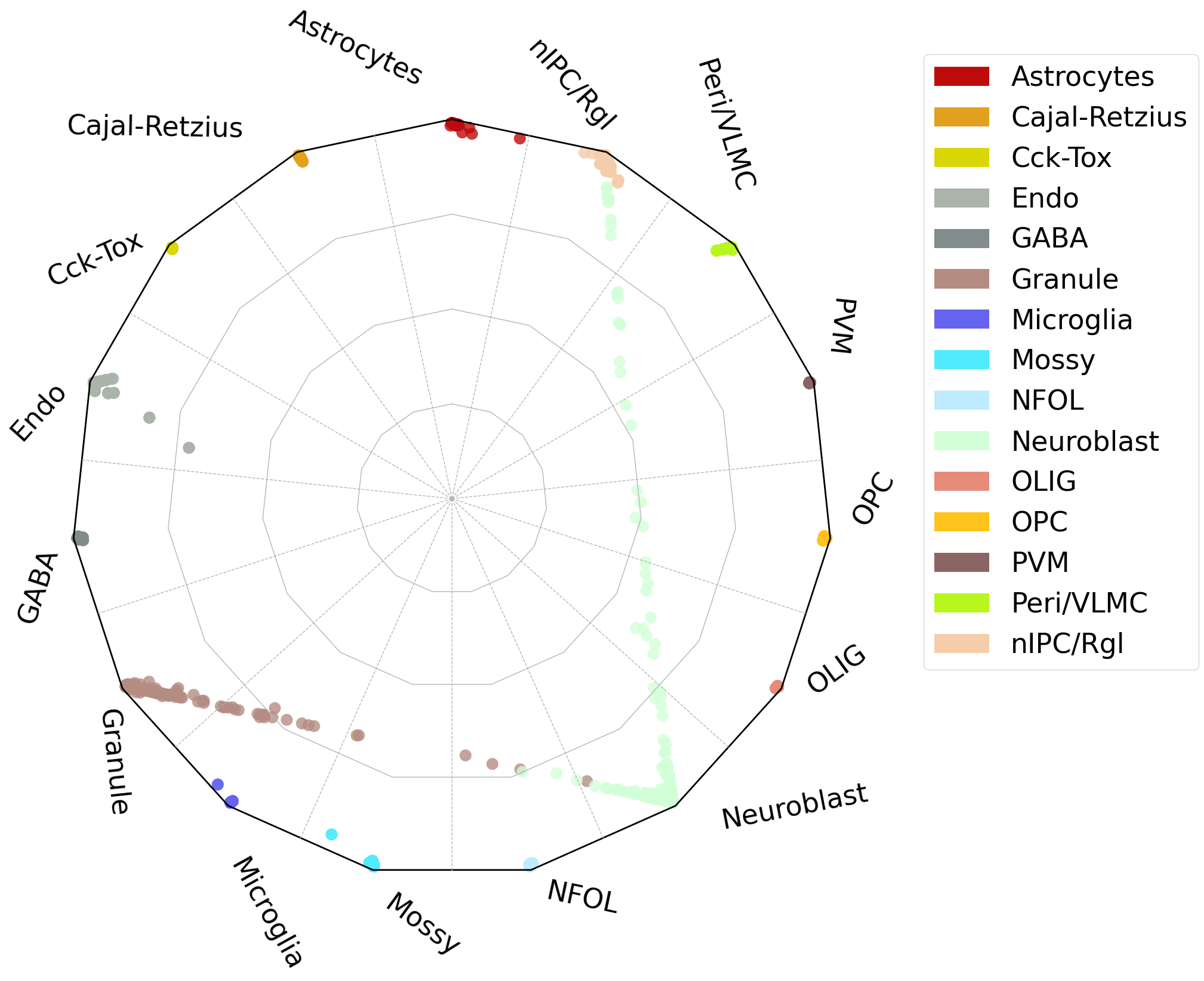
#radar plot
scref=scm.CamelSwapline.prediction(datax=scref, mcolor_dict=scref.uns["refcolor_dict"] ,net=net3,learninggroup="train", radarplot=True,fontsizeValue=10,
ncolnm=3, bbValue=(1.5, 1.55) )
#plt.savefig("upload_%s_RadarPlot_cluster.pdf"%today,bbox_inches='tight')
permutation control
[41]:
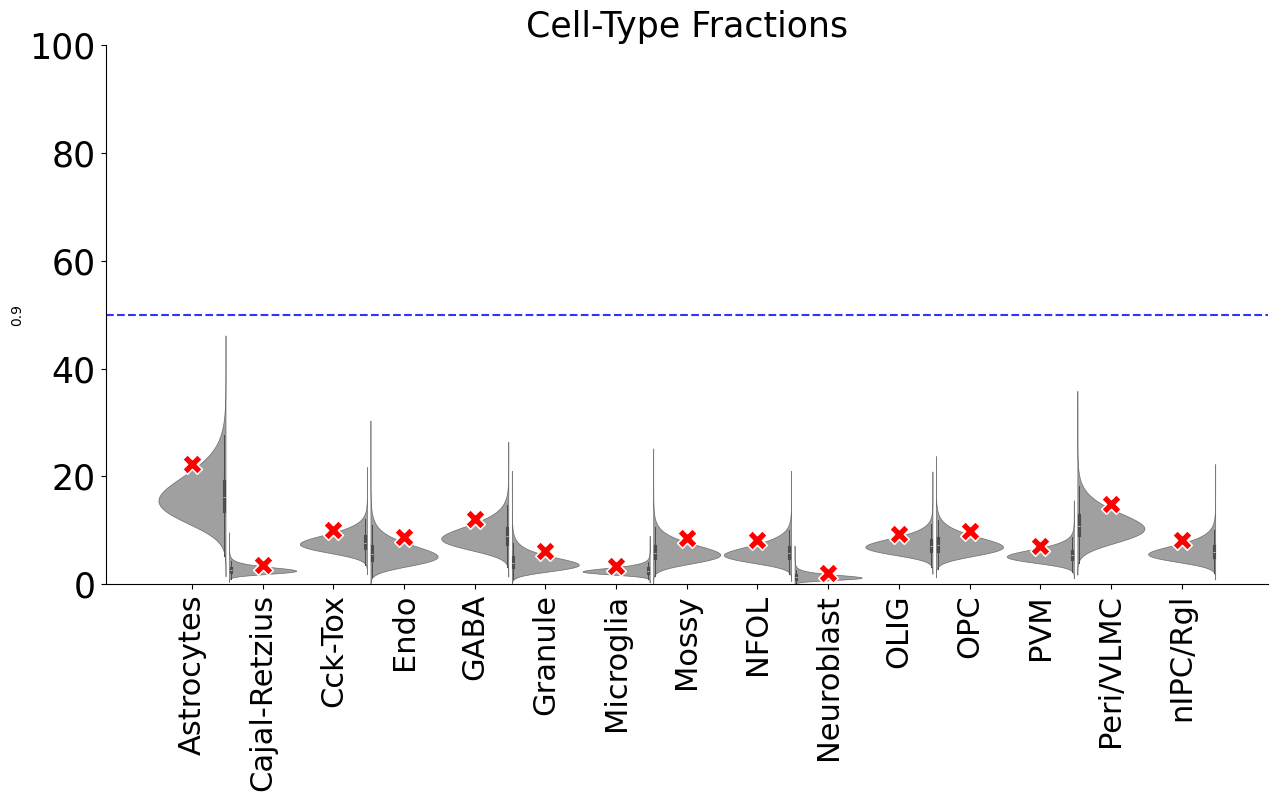
## the whole data matrix is randomized, the red X represents 95% conficence of each cell-type
[42]:
dftest0, ratiodf=scm.CamelSwapline.permutationTest(datax=scref,net=net,num=20, plotshow=True)
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: UserWarning:
The palette list has fewer values (1) than needed (15) and will cycle, which may produce an uninterpretable plot.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: FutureWarning:
The `scale_hue` parameter has been replaced and will be removed in v0.15.0. Pass `common_norm=True` for the same effect.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: FutureWarning:
The `bw` parameter is deprecated in favor of `bw_method`/`bw_adjust`.
Setting `bw_method=0.4`, but please see docs for the new parameters
and update your code. This will become an error in seaborn v0.15.0.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
<Figure size 640x480 with 0 Axes>
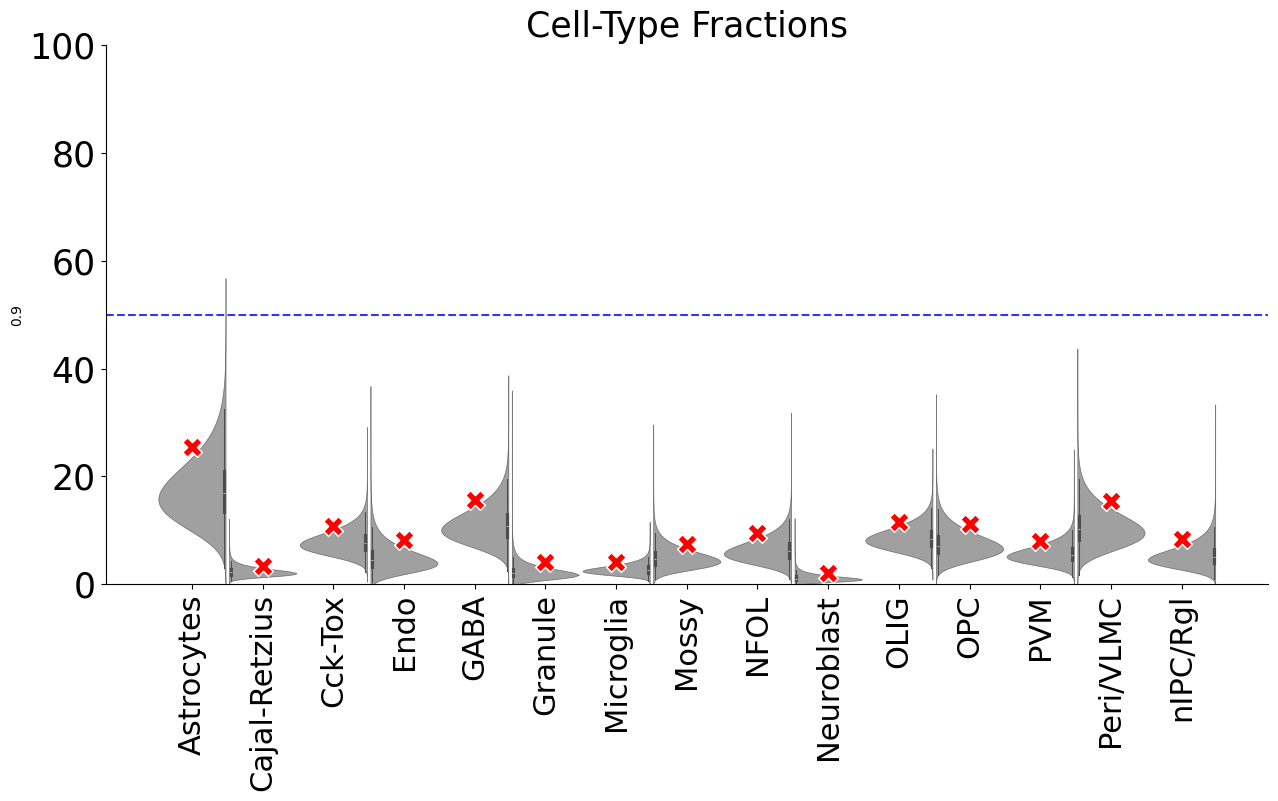
[43]:
dftest0, ratiodf=scm.CamelSwapline.permutationTest(datax=scref,net=net3,num=20, plotshow=True)
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: UserWarning:
The palette list has fewer values (1) than needed (15) and will cycle, which may produce an uninterpretable plot.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: FutureWarning:
The `scale` parameter has been renamed and will be removed in v0.15.0. Pass `density_norm='width'` for the same effect.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: FutureWarning:
The `scale_hue` parameter has been replaced and will be removed in v0.15.0. Pass `common_norm=True` for the same effect.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:1848: FutureWarning:
The `bw` parameter is deprecated in favor of `bw_method`/`bw_adjust`.
Setting `bw_method=0.4`, but please see docs for the new parameters
and update your code. This will become an error in seaborn v0.15.0.
ax = sns.violinplot(scale="width", bw=0.4, cut=2, gridsize=100, saturation=0.9, scale_hue=False,
<Figure size 640x480 with 0 Axes>
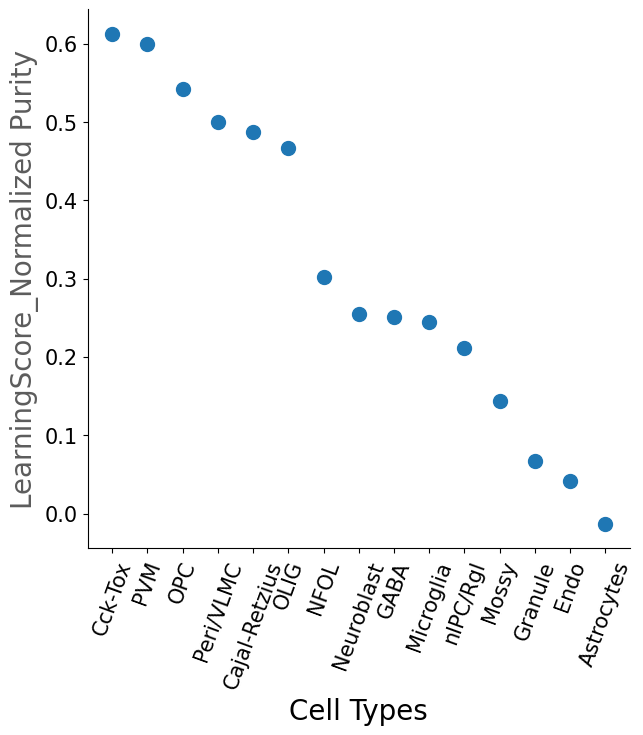
Cell_Type Purity
[44]:
#The ratio of the purity entropy for each cluster based on their learning scores, is used as a measure of purity.
#The function returns a pandas dataframe sorted by the purity score
[45]:
dfpurity1=scm.CamelSwapline.PurityEstimationLearningScore(datax=scref, clusterlist="Cluster", elbow=False, figureplot=True)
<Figure size 640x480 with 0 Axes>
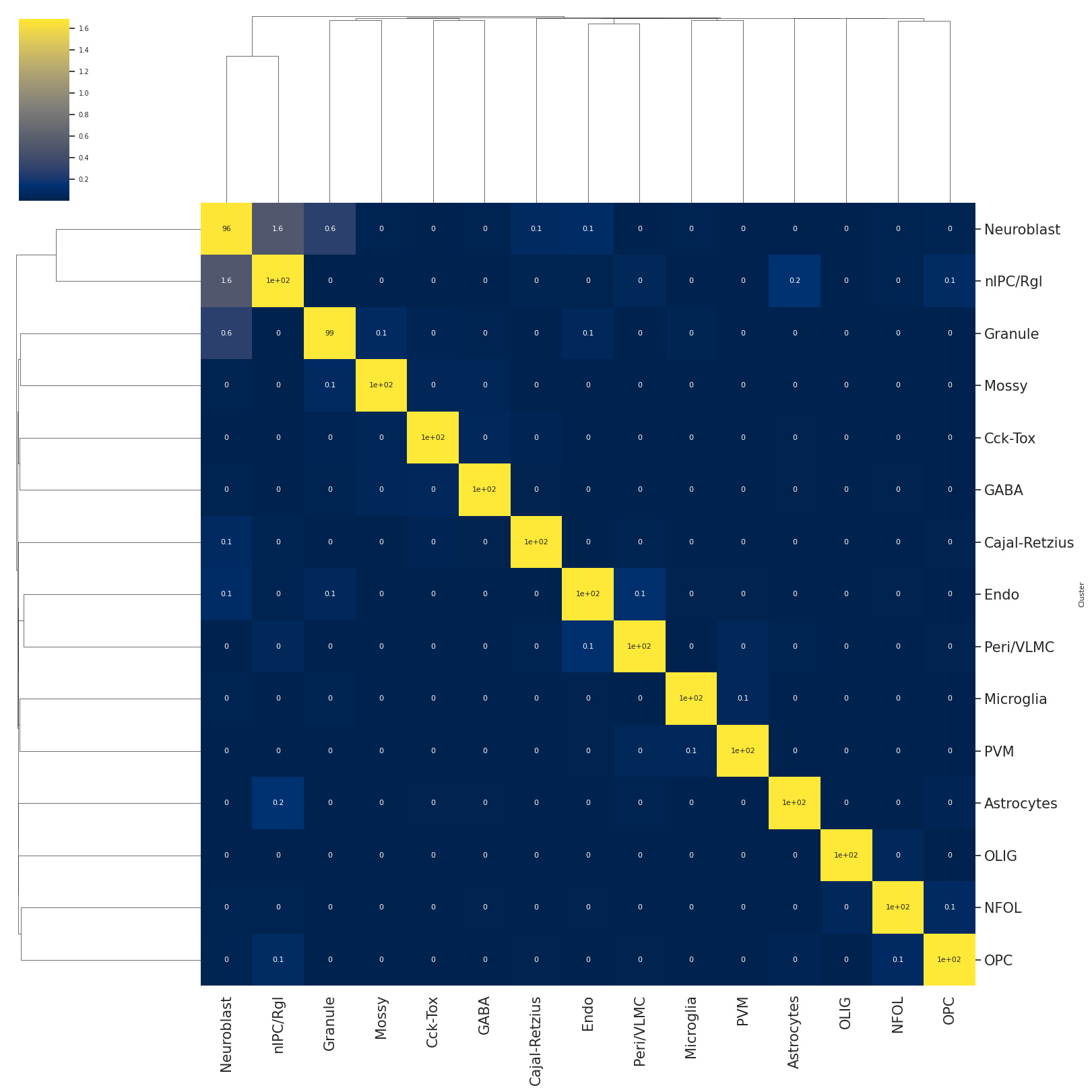
association between cell-types
[46]:
scref
[46]:
AnnData object with n_obs × n_vars = 13371 × 12516
obs: 'Cluster', 'Color', 'upsampled', 'mtrain_index', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'train_set_gene', 'mclasses_names', 'refcolor_dict', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'train_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[47]:
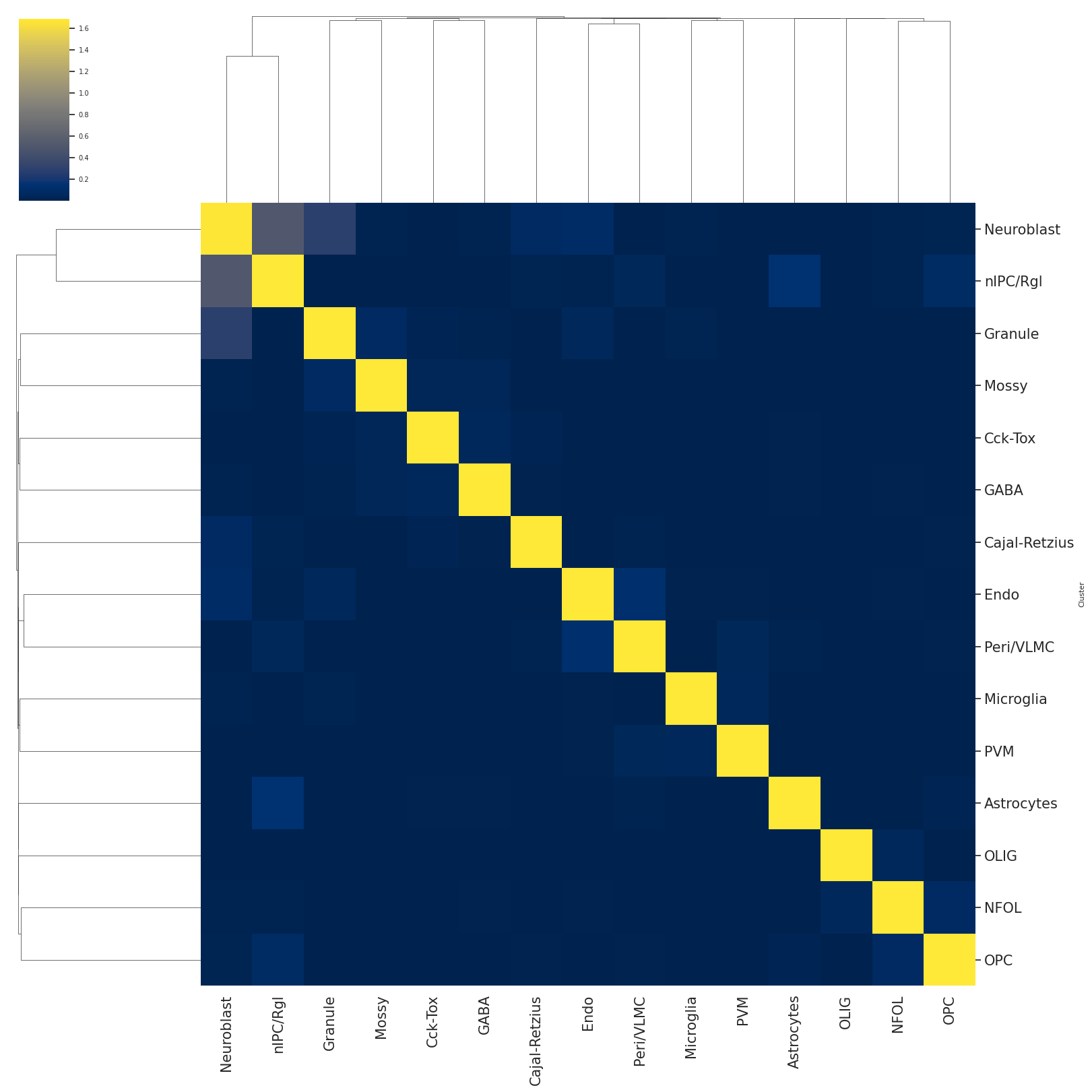
# the heatmap of hierarchical clustering represents the cell-type similarity or association
#color from dark purple to light yellow represents the association from low to high
#number inside of eahc square indicating the association value.
[48]:
scm.CamelSwapline.CellTypeSimilarity(datax=scref, labelnum=True, metricvalue='correlation',methodvalue="complete")
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:2472: FutureWarning: The default of observed=False is deprecated and will be changed to True in a future version of pandas. Pass observed=False to retain current behavior or observed=True to adopt the future default and silence this warning.
dfpb2 = dfprob.groupby(["Cluster"]).mean()
<Figure size 1500x1500 with 0 Axes>
[49]:
scm.CamelSwapline.CellTypeSimilarity(datax=scref, labelnum=False, metricvalue='correlation',methodvalue="complete")
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:2472: FutureWarning: The default of observed=False is deprecated and will be changed to True in a future version of pandas. Pass observed=False to retain current behavior or observed=True to adopt the future default and silence this warning.
dfpb2 = dfprob.groupby(["Cluster"]).mean()
<Figure size 1500x1500 with 0 Axes>
[ ]:
Save data
[50]:
scref
[50]:
AnnData object with n_obs × n_vars = 13371 × 12516
obs: 'Cluster', 'Color', 'upsampled', 'mtrain_index', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'train_set_gene', 'mclasses_names', 'refcolor_dict', 'mwanted_order', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'train_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[51]:
Path.cwd()
[51]:
PosixPath('/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse')
[52]:
scref.uns["refcolor_dict"]= predefined_colors
[53]:
work_dir=str(OUTPUT_DIR)
QueryName="ZeiselMouseDG"
TrainingName="ZeiselMouseDG"
filename="%s_%s_Ref%s_MergeCluster.h5ad"%(QueryName,TrainingName,today)
[54]:
os.path.join(work_dir,filename)
[54]:
'/mnt/e/Loal_Temp/Vicuna_Example/scCAMEL_VICUNA_updated_20260605/outputs/swapline_pypi047b0_tutorial/ZeiselMouseDG_ZeiselMouseDG_Ref2026-06-09_MergeCluster.h5ad'
[55]:
del scref.uns['refcolor_dict']
[56]:
CamelSwapline.write_data(adatax=scref,filename=filename,filepath=work_dir)
[57]:
#if color is not defined: scref.obs[ 'color']
scref.uns['refcolor_dict'] = pd.Series({
'Astrocytes': [190, 10, 10],'Cajal-Retzius': [225, 160, 30],'Cck-Tox': [217, 215, 7],
'Endo': [170, 180, 170], 'GABA': [130, 140, 140],'Granule': [180, 140, 130],
'Microglia': [100, 100, 240],'Mossy': [ 80, 235, 255],'NFOL':[190, 235, 255],
'Neuroblast':[210, 255, 215],'OLIG':[230, 140, 120], 'OPC': [255, 195, 28],
'PVM': [139, 101, 100],'Pericytes': [252, 183, 26],'Radial Glia-like': [214, 194, 39],
'VLMC': [255, 120, 155],'nIPC': [250, 145, 45],'hRgl2a': [250, 125, 25],
'hDA0': [190, 200, 190],'hOPC': [255, 35, 155],'hRN': [199, 121, 41],
'hNbGaba': [ 40, 55, 130],'hGaba': [ 7, 121, 61],'hOMTN': [ 95, 186, 70],
'hSert': [ 50, 180, 180],'nIPC/Rgl': [245, 205, 170], 'Peri/VLMC': [185, 245, 30],
'eSCc':[205,205,220]
})
[ ]:
[58]:
os.chdir(PROJECT_ROOT)
Path.cwd()
[58]:
PosixPath('/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse')
[59]:
import scanpy as sc
[60]:
scref=sc.read(OUTPUT_DIR / filename)
scref
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/_core/anndata.py:1756: UserWarning: Observation names are not unique. To make them unique, call `.obs_names_make_unique`.
utils.warn_names_duplicates("obs")
[60]:
AnnData object with n_obs × n_vars = 13371 × 12516
obs: 'Cluster', 'Color', 'upsampled', 'mtrain_index', 'color'
var: 'Filter1', 'MVgene', 'RefGeneList'
uns: 'Celltype_OrderNumber', 'Celltype_Score_RefCellType', 'mclasses_names', 'mwanted_order', 'train_set_gene'
obsm: 'CelltypeScoreCoordinates', 'Celltype_Score', 'train_set_values'
[61]:
net3= scm.CamelSwapline.load_camel_model(checkpoint_dir=str(OUTPUT_DIR / "camel_checkpoints2"), prefix="camel_nn_mouseDG",dropoutVal=0.3, device="cpu")
Prediction
Couturier2020_humanGlioblastoma
[62]:
os.chdir(PROJECT_ROOT)
Path.cwd()
[62]:
PosixPath('/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse')
[63]:
scpdt=anndata.read(PROJECT_ROOT / "Couturier2020_DevGBM_Ref2023-05-27.h5ad")
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/__init__.py:42: FutureWarning: `anndata.read` is deprecated, use `anndata.read_h5ad` instead. `ad.read` will be removed in mid 2024.
warnings.warn(
[64]:
set(scpdt.obs["Cluster"])
[64]:
{'Astro', 'Mesenchymal', 'Neuronal', 'Oligo', 'Progenitor', 'Unassigned'}
[65]:
scpdt
[65]:
AnnData object with n_obs × n_vars = 18475 × 33660
obs: 'Patient', 'Cluster', 'Color'
[66]:
scpdt.var.index
[66]:
Index(['A1BG', 'A1BG-AS1', 'A1CF', 'A2M', 'A2M-AS1', 'A2ML1', 'A2ML1-AS1',
'A2ML1-AS2', 'A3GALT2', 'A4GALT',
...
'ZXDC', 'ZYG11A', 'ZYG11B', 'ZYX', 'ZZEF1', 'ZZZ3', 'bP-21264C1.2',
'bP-2171C21.3', 'bP-2189O9.3', 'hsa-mir-1253'],
dtype='object', length=33660)
[67]:
scpdt.X=scpdt.X.todense()
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/anndata/_core/storage.py:39: ImplicitModificationWarning: X should not be a np.matrix, use np.ndarray instead.
warnings.warn(msg, ImplicitModificationWarning)
[68]:
scpdt2=scpdt.copy()
scpdt2=scm.CamelPrefiltering.DataScaling(scpdt2)
[69]:
########################################################
########################################################
#remeber to change the file path in tftable
########################################################
########################################################
scpdt =scm.CamelPrefiltering.MVgene_Scaling(datax=scpdt2,TPTT=0, commongene=scref.var.index.tolist(),
sharedMVgenes=scref.uns[ 'train_set_gene'].tolist(),
std_scaling=True,score=None, thrs=None, mprotogruop=None,
tftable=str(PUBLIC_DATASET / "FantomTF2CLUSTER_human_official.txt"), learninggroup="test")
scpdt.uns["mwanted_order"] =list(sort(list(set(scpdt.obs["Cluster"]))))
CamelRunning---GenesScaling......
CamelRunning---TestGenesScaling......Finished
[70]:
scpdt
[70]:
AnnData object with n_obs × n_vars = 18475 × 33660
obs: 'Patient', 'Cluster', 'Color', 'mtrain_index'
var: 'RefGeneList'
uns: 'train_set_gene', 'mclasses_names', 'mwanted_order'
obsm: 'test_set_values'
[71]:
#del scpdt.obs["color"]
[72]:
# if color is not definedi
#scpdt=scm.CamelSwapline.addcolor(datax=scpdt,clustername="Cluster", colorcode="color")
[19]:
[73]:
scpdt.uns["refcolor_dict"] = pd.Series({'Astro': [100, 100, 240], 'Neuronal': [ 0, 86, 255],
'Mesenchymal': [55, 120, 55], 'Oligo': [ 255,185, 5], 'Unassigned': [192,192,192],
'Progenitor': [190, 0, 0]})
[74]:
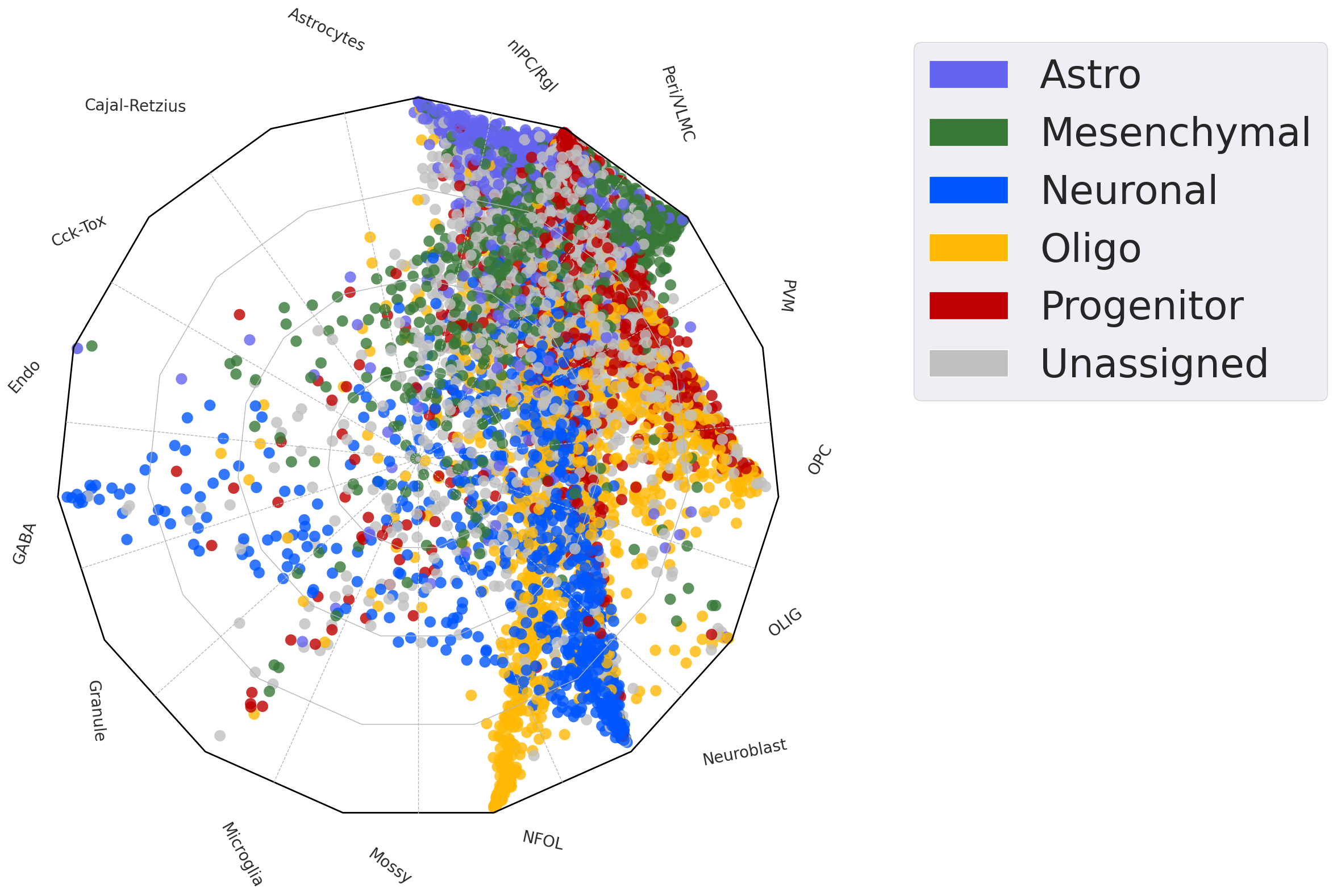
test=scm.CamelSwapline.prediction(datax=scpdt, mcolor_dict=pd.Series(scpdt.uns["refcolor_dict"]),net=net3,
learninggroup="test", radarplot=True, fontsizeValue=35,
datarefplot=scref,ncolnm=1, bbValue=(1.1, 1.05))
[75]:
scpdt
[75]:
AnnData object with n_obs × n_vars = 18475 × 33660
obs: 'Patient', 'Cluster', 'Color', 'mtrain_index'
var: 'RefGeneList'
uns: 'train_set_gene', 'mclasses_names', 'mwanted_order', 'refcolor_dict', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'test_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[76]:
genename=sort(list(set(scpdt.obs["Cluster"])))
name=sort(list(set(scref.obs["Cluster"])))
[77]:
dfprob=pd.DataFrame(scpdt.obsm['Celltype_Score'])
dfprob.columns=scpdt.uns['Celltype_Score_RefCellType']
dfprob.index=scpdt.obs.index
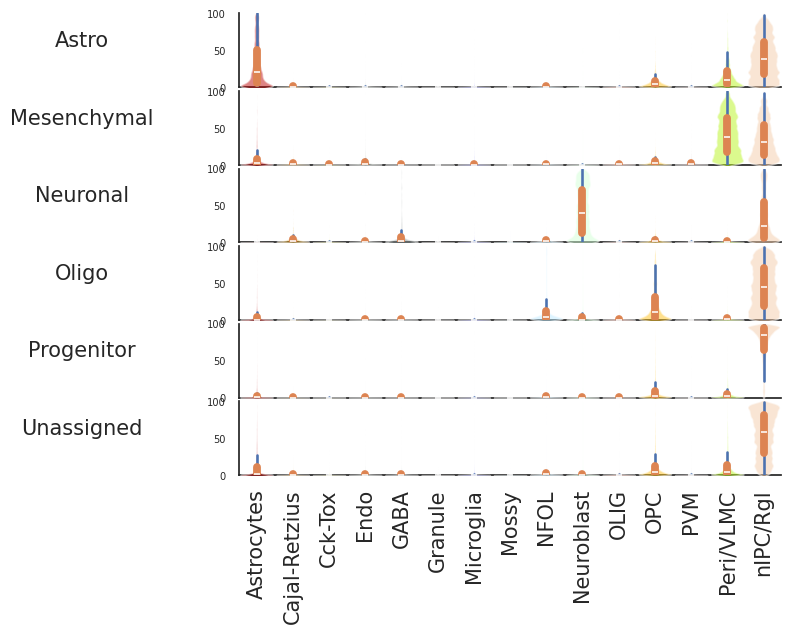
dfmk=dfprob.astype(float).join(scpdt.obs["Cluster"],how="inner").T
dfprob=CamelSwapline.CellTypeSimilarityViolinPlot(datax=scpdt, dataref=scref)
/home/huyiz/anaconda3/envs/py310/lib/python3.10/site-packages/scCAMEL/CamelSwapline.py:883: UserWarning: This figure includes Axes that are not compatible with tight_layout, so results might be incorrect.
fig.tight_layout()
Save data
[78]:
Path.cwd()
[78]:
PosixPath('/mnt/e/YZstudio/OneDrive/Research/Dataset/Brain_Adult_mouse')
[79]:
scpdt
[79]:
AnnData object with n_obs × n_vars = 18475 × 33660
obs: 'Patient', 'Cluster', 'Color', 'mtrain_index'
var: 'RefGeneList'
uns: 'train_set_gene', 'mclasses_names', 'mwanted_order', 'refcolor_dict', 'Celltype_Score_RefCellType', 'Celltype_OrderNumber'
obsm: 'test_set_values', 'Celltype_Score', 'CelltypeScoreCoordinates'
[80]:
work_dir=str(OUTPUT_DIR)
QueryName="Couturier2020"
TrainingName="ZeiselMouseDG"
filename="%s_%s_Ref%s_MergeCluster.h5ad"%(QueryName,TrainingName,today)
[81]:
os.path.join(work_dir,filename)
[81]:
'/mnt/e/Loal_Temp/Vicuna_Example/scCAMEL_VICUNA_updated_20260605/outputs/swapline_pypi047b0_tutorial/Couturier2020_ZeiselMouseDG_Ref2026-06-09_MergeCluster.h5ad'
[82]:
del scpdt.uns["refcolor_dict"]
[83]:
CamelSwapline.write_data(adatax=scpdt,filename=filename,filepath=work_dir)
[ ]:
[ ]:
[ ]:
[ ]:
[ ]:
[ ]: